BEYAZ HÜCRE TÜMÖRLERİ
Etyoloji ve patogenez
Kromozomal translokasyon ve onkogenler:
B lenfositlerinde daha sık
Herediter genetik faktörler:
Bloom sendromu, Fankoni anemisi ve ataksi telenjiektazi, Down sendromu, tip I Nörofibromatozis
Virüsler:
HTLV-1 :Erişkin T hücreli lösemi ve lenfoması
EBV: Burkitt lenfoma, Hodgkin lenfoma olgularının %30-40 kadarı, immünyetmezlikte görülen B hücreli lenfomalar ve nadir NK hücreli lenfomalar
KSHV/HHV-8: Sıklıkla plevral kavitede maliğn effüzyon şeklinde görülen B hücreli lenfoma
Çevresel etkenler:
Helikobakter pilori enfeksiyonu, gastrik B hücreli lenfoma
Gluten enteropatisi T hücreli intestinal lenfoma
HIV enfeksiyonunda B hücreli lenfoma ve EBV’ a bağlı lenfomalar
İatrojenik faktörler:
Radyoterapi ve bazı kemoterapi ajanları
WHO Klasifikasyonu
|
1) Prekürsör B hücre tümörleri |
4) Periferik T ve NK hücre tümörleri |
|
Prekürsör B lenfoblastik lösemi/lenfoma |
T hücre prolenfositik lösemi Büyük granüler lenfositik lösemi Mikozis fungoides/Sezary sendromu Periferal T hücreli lenfoma, nonspesifik Anaplastik büyük hücreli lenfoma Anjioimmünoblastik T hücreli lenfoma Enteropati ile ilişkili T hücreli lenfoma Pannikülit benzeri T hücreli lenfoma Hepatosplenik T hücreli lenfoma Erişkin T hücreli lösemi/lenfoma NK/T hücreli lenfoma, nazal tip NK hücreli lösemi |
|
2) Periferik B hücre tümörleri |
5) Hodgkin Lenfoma |
|
Kronik lenfositik lösemi/ küçük lenfositik lenfoma B hücre prolenfositik lösemi Lenfoplazmositik lenfoma Splenik ve nodal marjinal zon lenfoma Ekstranodal marjinal zon lenfoma Mantle hücreli lenfoma Foliküler lenfoma Marjinal zon lenfoma Hairy cell lösemi Plazmositom/plazma hücreli myelom Diffüz büyük B hücreli lenfoma Burkitt lenfoma |
Klasik tipler · Nodüler skleroz · Karışık hücreli · Lenfositten zengin · Lenfositten fakir Lenfosit baskın |
|
3) Prekürsör T hücre tümörleri |
|
|
Prekürsör T lenfoblastik lösemi/ lenfoma |
Akut lenfoblastik Lösemi/Lenfoma
%85 prekürsör B hücrelerinden oluşan, yaygın kemik iliği ve değişken periferik kan tutulumu gösteren çocukluk çağı akut lösemileridir. Prekürsör T hücreli olanlar daha az görülüp, çoğu adolesan erkeklerde timik tutulum ile karakterli lenfoma şeklindedir. Olguların çoğu 15 yaş altında olup beyaz ırkta ve erkek çocuklarında biraz daha sık görülür.
• Morfoloji: Normal doku yapısını bozan lenfoblast infiltrasyonu, bazı hücrelerde çentikli görünüm, yüksek mitoz oranı, ölen hücreli temizlemek için gelen makrofajlarla oluşan yıldızlı gök manzarası benzeri görünüm
• İmmünfenotip: pre-B ve pre-T hücrelerinde bulunan TdT, >%95 olguda bulunur. T ve B hücre ayırımı için spesifik belirleyiciler kullanılır. Pre-B hücresi CD19, CD10; pre-T hücresi CD1, CD2, CD5 ve CD7.
• Kötü prognoz bulguları: 2 yaş altı; adolesan ve erişkin yaş; periferik kanda 100,000 üzerinde blastik hücre; çocukluk çağında %3, erişkinde %25 olguda belirlenen t(9;22) – Philadelphia kromozomu
• İyi prognoz bulguları: 2-10 yaş; düşük sayıda hücre, erken pre-B fenotipi, hiperploidi yada t(12;21)
PERİFERİK B HÜCRE TÜMÖRLERİ
Kronik lenfositik lösemi (CLL)/ Küçük lenfositik lenfoma (SLL)
Bu iki hastalık morfolojik, fenotipik ve genotipik olarak birbirinden ayrılamaz. Aralarındaki tek farklılık, periferik kandaki lenfositoz derecesidir. Çoğu hastada lenfosit sayımı >4000/mm3 olup lösemi tanısı alır. Batılı ülkelerde en sık görülen erişkin çağ lösemisidir.
• Morfoloji: Lenf nodunu diffüz infilte eden yuvarlak, hafif düzensiz nüveli, dar sitoplazmali küçük lenfositler; lösemik formunda kanda bulunan lenfositler frajil olup yayma hazırlanırken ezilmiş hücreler
• İmmünfenotip: CD19, CD20, CD23 ve CD5 düşük düzeyde yüzey immünglobülin ekspresyonu (IgM/ IgM ve IgD)
• Klinik: Çoğu 50 yaş üzeri (median yaş 60) erkek hastalardır (M/F: 2/1). Çoğu asemptomatiktir. Jeneralize lenfadenopati, hepatosplenomegali %50-60 olguda mevcuttur. Kemik iliği tutulumu olan SLL olgularında lökopeni; ağır CLL olgularında >200,000/mm3 üzeri periferik lökositoz olabilir. Hipogamaglobülinemi sıktır. %10-15 olguda otoimmün hemolitik anemi yada trombositopeni; ortalama sağkalım 4-6 yıl; %10 olguda diffüz büyük B hücreli lenfomaya transformasyon (Richter sendromu)
Foliküler Lenfoma
ABD’ de en sık görülen NHL olup erişkin lenfomalarının %45’ ini oluşturur. En sık orta yaşta; kadın ve erkekde aynı oranda görülür. Neoplastik hücreler normal germinal merkez B hücrelerine benzer.
• Morfoloji: Lenf nodunda nodüler patern; çoğunda küçük çentikli hücreler baskın; %85 olguda kemik iliği tutulumu; lösemik dönüşüm nadir
• İmmünfenotip: CD19, CD20, CD10, yüzey immünglobülinleri BCL2 protein ekspresyonu (normal foliküler hücrelerinden farkı)
• Sitogenetik ve moleküler genetik: %90 olguda t(14;18) sonucu IgH (ağır zincir) lokusu ile 18. kromozomdaki BCL2 lokusu birleşir ve BCL2 ekspresyonu artar. Apoptoz engellenir ve neoplastik B hücreleri çoğalır.
• Klinik: Ağrısız, jeneralize lenfadenopati; ortalama sağkalım 7-9 yıl; tedaviye yanıt yok; %30-50 olguda diffüz büyük B hücrei lenfomaya transformasyon
Diffüz Büyük B Hücreli Lenfoma
Tüm NHL olgularının %20; agresif lenfomaların %60-70 kadarını oluşturur. En sık 60 yaş civarında ve erkeklerde biraz daha sık görülür. Çocukluk çağı lenfomalarının %5 kadarını oluşturur.
• Morfoloji: Büyük boyutlu lenfositler ve diffüz büyüme paterni
• İmmünfenotip: CD19, CD20, bazılarında CD10 ve BCL6, çoğunda yüzey Ig
• Onkogenik virüsler ile ilişkili subtipler: Ağır T hücre yetmezliğinde (ileri evre HIV enfeksiyonu, ağır kombine immün yetmezlik, transplantasyon) EBV etkisi; ileri evre HIV enfeksiyonunda görülen vücut kavite lenfomalarında KSHV/HHV8 etkisi
• Klinik ve prognoz: Tek bir nodal yada ekstranodal bölgede, hızla büyüyen semptomatik kitle; kemik iliği geç dönemde tutulur ve lösemik tablo nadir; tedavi edilmezse kötü prognoz; yoğun kombine kemoterapi ile %60-80 komplet remisyon ve yaklaşık %50 olguda iyileşme
Burkitt Lenfoma
• Morfoloji: Orta boyutlu, birkaç nükleolü olan nüveli hücreler; sık mitoz ve apoptoz; kalıntıları fagosite eden makrofajlar nedeni ile yıldızlı gök manzarası
• İmmünfenotip: Yüzey IgM, monotipik hafif zincirler, CD19, CD20, CD10 ve BCL6 gibi matür B hücre belirleyicileri
• Sitogenetik ve moleküler genetik: Tümünde 8. kromozom da yerleşen c-MYC geninde translokasyon t(8;14).
• Endemik tümörlerin tümünde; AIDS hastalarının %25’i, sporadik olguların %15-20’ sinde latent EBV enfeksiyonu
• Klinik: Endemik ve sporadik olguların çoğu çocuk ve genç erişkinlerde görülür. ABD’ de çocukluk çağı lenfomalarının %30’ u Burkitt lenfomadır. Çoğu ekstranodal yerleşir. Endemik olanlar mandibula yanı sıra böbrek, over ve adrenal gland gibi abdominal organlarda yerleşir. Sporadik olgular daha sık olarak ileoçekal bölge ve peritonda yerleşir. Kemik iliği ve periferik kan tutulumu nadirdir. Çok agresif olmakla birlikte, kısa süreli, yüksek doz kemoterapiye iyi yanıt verir. Çoğu çocuk ve genç erişkin iyileşirken, yaşlılarda kötü davranış gösterir.
*************Çocuklarda görülen non-Hodgkin lenfomalar agresif tümörler olup kötü prognozludur. Ama hücreler hızlı çoğaldığından kemoterapiye iyi yanıt verirler ve erken tanı ile iyileşme şansları yüksektir. Burkitt lenfoma vücutta en hızlı çoğalan tümör olarak bildirilmektedir***********************
PLAZMA HÜCRE TÜMÖRLERİ
• Multiple myelom (plazma hücre myelomu) En önemli ve en sık semptomatik monoklonal gamopatidir. İskelet sisteminde yaygın odaklar yapar. Nadir görülen soliter myelom yada plazmositom ise kemik yada bazı yumuşak dokularda tek (soliter) kitle yapar.
• Waldenström makroglobülinemisi: IgM düzeyi yüksekiliğine bağlı gelişen hipervizkozite vardır. Lenfoplazmositik lenfoması olan erişkinlerde görülür.
• Ağır zincir hastalığı: Serbest H zincirleri üretip salgılayan bir gruptur. CLL/SLL, lenfoplazmositik lenfoma ve ince barsakta nadir görülen bir tümöral proliferasyon (Akdeniz lenfoması) bu tabloya yol açabilir.
• Primer yada immünosit ile ilişkili amiloidoz: serbest L zincirleri üreten plazma hücre proliferasyonu olup amiloidoz ile sonuçlanır. Bazı hastalarda belirgin multiple myelom; bazılarında ise yalnızca kemik iliğinde minör klonal plazma hücre proliferasyonu belirlenir.
• Önemi belirlenemeyen monoklonal gamopati: Klinik bulgusu olamayan hastalarda kanda m komponentlerinin belirlenmesidir. Yaşlılarda oldukça sık olmakla birlikte semptomatik olgulara (en sık multiple myelom) dönüşüm nadirdir.
Multiple Myelom
• İskelet sisteminde plazma hücrelerinden oluşan multifokal, destrüktif tümör odakları; sıklık sırasına göre vertebra, kotsalar, kafa tası, pelvis, femur, klavikula ve skapula; radyolojik olarak 1-4 cm çapında, zımba ile delinmiş görünümde lezyonlar
• Düzensiz immünglobülin üretimine bağlı hücre içi birikimler değişik görünümlere yol açar: alev kırmızısı sitoplazma (alev hücreleri); mavi görünümde üzüm tanesi benzeri sitoplazmik damlacıklar (Mott hücreleri); sitoplazmik Russel cisimcikleri; intranükleer Dutcher cisimcikleri;
• Yüksek düzeyde M proteini nedeni ile eritrositlerin birbirine yapışması: rulo formasyonu
• Enfeksiyonlardan sonra, en sık ölüm sebebi böbrek yetmezliği
• %6-15 hastada amiloidoz
• Kesin tanı kemik iliği incelemesi ile
PERİFERİK T VE NK HÜCRE TÜMÖRLERİ
Erişkin T Hücreli Lösemi/Lenfoma
• CD4+ T lenfositlerinin tümörü olup HTLV-1 virüsü ile ilişkili
• Japonya, batı Afrika ve Karaib adaları gibi, virüsün endemik olarak bulunduğu yerlerde daha sık
• Cilt lezyonları, jeneralize lenfadenopati, hepatosplenomegali, periferik lenfositoz ve hiperkalsemi
• Tutulan bölgeler ve periferik kanda multilobe nüveli (çiçek hücreleri) hücreler
Mikozis Fungoides/Sezary Sendromu
• CD4+ T lenfositlerinin maliğn tümörü olup cilt tutulumu ön planda
• Epidermis ve üst dermisi infiltre eden maliğn hücrelerda serebriform nüveler
• Ekstrakutanöz yayılım lenf nodu ve kemik iliğine
• Sezary sendromunda jeneralize eksfoliatif eritrodermi ve eşlik eden Sezary (serebriform) hücrelerini içeren lösemi
• Yavaş gidişli olup ortalama sağkalım 8-9 yıl
************Non-Hodgkin lenfomaların bir kısmında lösemik dönüşüm sıktır. Bu grupta lösemi ve lenfoma hücreleri aynı genotipik, fenotipik ve morfolojik özelliklere sahiptir. Bu tümörler: küçük hücreli lenfoma/kronik lenfositik lösemi; T hücreli lenfoblastik lenfoma/T hücreli ALL; mikozis fungoides/Sezary sendromu****************
HODGKİN LENFOMA
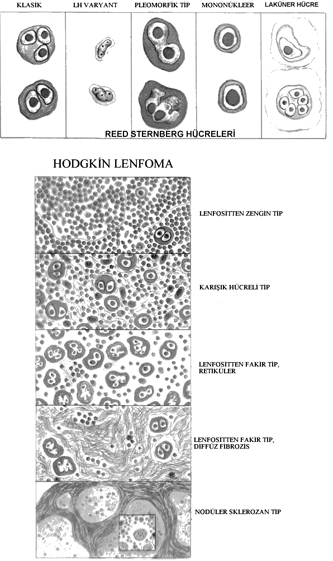
Primer olarak lenfoid dokuları tutan bir hastalıktır. Tek bir lenf nodu veya nod zincirinde gelişip
ilişkili olduğu lenf nodlarına yayılır.
• Reed-Sternberg (RS) denilen neoplastik dev hücreler
• Ateş gibi belirgin klinik bulgular ile NHL’dan ayrılır
Reed- Sternberg hücreleri germinal merkez yada post-germinal merkez B hücrelerinden köken alırlar. Bu nedenle Hodgkin lenfoma B hücre tümörü olarak kabul edilmektedir. Genç erişkinlerde en sık görülen maliğn tümörlerden biri olup tanı anında ortalama yaş 32 dir. Tedavide gelişmeler sonucunda çoğu olguda iyileşme sağlanmaktadır.
WHO sınıflaması:
• Nodüler sklerozis
• Karışık hücreli
• Lenfositten zengin
• Lenfositten fakir
• Lenfosit baskın
***************İlk dört tipinde (klasik tipler) Reed-Sternberg hücrelerinde CD20 negatif; CD15 ve CD30 pozitiftir. Lenfosit baskın tipte lenfohistiositik varyant (popkorn) hücrelerde CD20 pozitif; CD15 ve CD30 negatiftir.***************************
• Nodüler sklerozis tipi: En sık görülen tip (%65-70). Laküner hücreler ve lenf nodunu nodüllere ayıran kollajen bantlar görülür. F=M. Alt servikal, supraklaviküler ve mediastinal lenf nodu tutulumu sıktır. Ergenlik ve genç erişkinlerde sık olup nadiren EBV ile ilişkilidir. Prognozu mükemmeldir.
• Karışık hücreli tip: %20-25 olguyu oluşturur. Küçük T lenfositi, eozinofil, plazma hücresi ve beniğn makrofajlardan oluşan hücrelerin arasında diagnostik ve mononükleer Reed-Sternberg hücreleri görülür. Erkeklerde daha sıktır. %70 kadar olguda EBV ile ilişkilidir. İleri yaşta, sistemik semptomlarla birlikte olup sıklıkla ileri evrededir. Yine de prognoz çok iyidir.
• Lenfositten zengin tip: Nadirdir. Mononükleer ve diagnostik Reed-sternberg hücrelerinin sık olması ile lenfosit baskın tipten ayrılır. %40 olguda EBV ile ilişkilidir. Prognozu çok iyi yada mükemmeldir.
• Lenfositten fakir tip: En az görülen tiptir. Lenfositler az olup Reed-Sternberg hücreleri ve pleomorfik tipleri sıktır. Büyük hücreli NHL ile karışır. Yaşlılarda daha sık görülür. HIV (+) kişilerde, geri kalmış toplumlarda daha sık olup sıklıkla EBV ile ilişkilidir. İleri evre ve sistemik bulgular sık olup diğer tiplere göre kötü prognozludur.
• Lenfosit baskın tip: Nadirdir. Olguların %5 kadarını oluşturur. Beniğn histiositler ile karışık küçük lenfosit infiltrasyonu görülür. Tipik Reed- Sternberg hücreleri azdır. Lenfohistiositik varyant (popkorn hücre) sıktır. Eozinofil, plazma hücresi ve nötrofiller az olup nekroz ve fibrozis izlenmez. EBV ile ilişkili değildir. Hastaların çoğu 35 yaş altı, erkeklerdir. Servikal yada aksiller lenfadenopati sık olup mediasten yada kemik iliği tutulumu nadirdir. Tekrarlama riski olsa da, prognoz mükemmeldir.
LANGERHANS HÜCRELİ HİSTİOSİTOZ
Dendritik yapıdaki Langerhans hücreleri HLA-DR, S-100 ve CD1a eksprese ederler. Sitoplazmada Birbeck granülleri karakteristiktir.
Multifokal multisistem Langerhans hücreli histiositoz (Letterer-Siwe hastalığı)
• En sık 2 yaş altında
• Seborreik döküntülere benzeyen cilt lezyonları, hepatosplenomegali, lenfadenopati, pulmoner lezyonlar ve sonunda destrüktif kemik lezyonları
• Kemik iliği infiltrasyonu pansitopeni, tekrarlayan otitis media ve mastoidit gibi enfeksiyonlar
• Tedavi edilmez ise öldürücüdür. Yoğun kemoterapi ile 5 yıllık sağkalım %50’ dir.
Unifokal unisistem Langerhans hücreli histiositoz (eozinofilik granülom)
• En sık kemik iliğini tutan, genişleyen eroziv lezyonlar
• Histiositler eozinofil, nötrofil, lenfosit ve plazma hücreleri ile karışık
• En sık kafa kemikleri, kotsalar ve femurda
• Asemptomatik olabilir yada ağrı, hassasiyet ve patolojik kırığa yol açar.
• Spontan olarak gerileyebileceği gibi, lokal eksizyon yada radyoterapi ile iyileşir.
•
Multifokal unisistem Langerhans hücreli histiositoz
• Genellikle küçük çocuklarda, multifokal, yumuşak dokuya uzanan, eroziv kemik defektleri
• Hastaların yaklaşık %50’sinde arka hipofiz sapı tutulumu sonucunda diabetes insipitus
• Kalvarial kemik defektleri, diabetes insipitus ve ekzoftalmus kombinasyonuna Hand-Schüller-Christian triadı denir.
• Çoğu hastada spontan gerileme;diğerlerinde kemoterapi ile tedavi
TİMUS
Hiperplazi : Normal timusta lenfoid foliküller yoktur. Hiperplazide medullada lenfoid foliküller oluşur. Myastenya gravis, SLE ve romatoid artrit gibi otoimmun hastalıklarda çoğunlukla timusda hiperplazi oluşur. Myastenya graviste timusun myoid hücrelerine karşı duyarlılık kazanan T hücreleri lenfoid foliküllerde B hücrelerini uyararak antikor oluşumuna yol açar. Bunun sonucunda nöromuskuler bileşkedeki asetil kolin reseptörlerine karşı otoimmun reaksiyon gelişir.
Timoma
Timustaki epitelyal hücrelerin tümörleridir. Tümörde bulunan lenfositler normal yapıda olup neoplazik değildir.
Beniğn Timoma : Sitolojik ve biolojik olarak beniğndir. Medulladaki epitel hücrelerine benzer hücreler oluşturur. Timomaların %60-70’ini oluşturur.
Malign Timoma : Tip I : Sitolojik olarak beniğn görünsede lokal invazyon ve nadiren uzak metastaz yapabilir. Timomaların %20-25’inin oluşturur.
Tip II (Timik Karsinom) :Hem sitolojik olarak hemde biolojik olarak maligndir. Timomaların %5’ini oluşturur. Çoğu iyi veya az diferansiye skuamöz karsinomdur. Daha az olarak görülen lenfoepitelyoma da olgun görünümlü lenfositlerde tümöre eşlik eder. Bir kısmında EBV varlığı belirlenmiştir. Bu açıdan nazofaringeal karsinoma benzer.
Timomalar orta yaşta sıktır. Myastenya gravisli olguların %15-20’sinde timoma bulunur. Tümör çıkarıldığında nöromuskuler bozukluk geriler. Hipogamaglobulinemi, SLE, saf eritrosit aplazisi ve nontimik kanserlerde timoma ile ilişkili olabilir.
DALAK
Primer tümörleri fibrom, osteom, kondrom ve en sık görülen lenfanjiom ile hemanjiom; maliğn olarak anjiosarkom, fibrosarkom.
Splenomegali :
Çoğu olguda splenomegali başka bir bölgedeki primer hastalığa sekonder olarak gelişir. Eritrosit, trombosit ve lökositler dalakta yıkılabilir.
İleri Derecede Splenomegali (1000 gm )
1. Kronik Myeloproliferatif hastalık (KML, Myeloid metaplazi)
2. KLL (daha az oranda)
3. Hairy cell lösemi
4. Sıtma
5. Gaucher hastalığı
6. Lenfomalar
7. Primer dalak tümörleri (nadir)
Orta Derecede Splenomegali (500-1000 gm)
1. Kronik konjestif splenomegali (portal hipertansiyon veya splenik ven obstrüksiyonu)
2. Akut lösemi
3. Herediter Sferositoz
4. Talasemi mayor
5. Otoimmun hemolitik anemi
6. Amiloidoz
7. Niemann-Pick hastalığı
8. Langerhans hücreli histiositoz
9. Kronik splenit (özellikle enfektif endokardit ile birlikte)
10. Tüberküloz, sarkoidoz, tifo
11. Metastatik karsinom veya sarkom
Hafif derecede Splenomegali ( 500 gr. altında)
1. Akut splenit
2. Akut splenik konjesyon
3. Enfeksiyöz mononükleoz
4. Septisemi, SLE, intra-abdominal enfeksiyonlar gibi akut febril hastalıklar.
Dalak büyümelerinde hipersplenizm oluşabilir anemi, lökopeni, trombositopeni
****************Anemi ve splenomegali ilişkisi sık olarak soruluyor. Herediter sferositoz, otoimmün hemolitik anemi ve talasemi major dışında kalan anemilerde belirgin splenomegali görülmez.**********