NEFROTİK SENDROM
- Masif proteinüri (3.5 gm/gün ↑)
- Yaygın ödem
- Hipoalbuminemi (3mg/dl↓)
- Hiperlipidemi – lipidüri
Başlangıçta azotemi, hematüri, hipertansiyon yoktur. Nefrotik sendrom 15 yaş altı çocuklarda genellikle primer, erişkinlerde ise sistemik hastalıkla ilişkilidir .
Primer olarak çocuklarda en sık nefrotik sendrom oluşturan lipoid nefrozdur (minimal lezyon hastalığı). Erişkinde nefrotik sendromda önce diabet, SLE veya amiloidoz gibi sistemik hastalıklar ekarte edilmelidir. Primer olarak erişkinde en sık nefrotik sendrom sebebi membranöz glomerulonefrittir.
Minimal lezyon hastalığı (lipoid nefroz)
Işık mikroskobu ile incelendiğinde normal yapıdan çok fazla farklılık göstermediğinden minimal değişiklik veya minimal lezyon hastalığı olarak isimlendirilmiştir.
- Işık mikroskobu ile glomerüller normaldir. Işık mikroskobu (IM) ile görülen tek patoloji zedelenmiş glomerülden süzülen lipoproteinlerin reabsorbsiyonu ile proksimal tübül hücrelerinde oluşan lipid birikimidir ve bu nedenle lipoid nefroz olarak da isimlendirilir.
- EM ile ise viseral epitel ayaksı çıkıntılarında diffüz kayıp, düzleşme görülür.
- İmmünfloresan mikroskobisinde birikim görülmez.
Bulgular geri dönüşümlüdür. 2-3 yaş arası en sık görülür. Patogenezi tam olarak bilinmesede atopi varlığı, viral enfeksiyon sonrası görülebilmesi, Hodkin lenfoma ile birlikte görülebilmesi, kortikosteroide iyi yanıt vermesi ve herhangi bir birikimin olmaması nedeni ile hücresel immünite bozukluğu (T lenfositi) ile ilişkili olduğu düşünülmektedir.
Olguların %90’ı kortikosteroide yanıt verir. Ama proteinüri tekrarlayabilir. Üçlü filtrasyon bariyerinin yalnızca tek elemanında defekt olduğundan (pedisellerde düzleşme) proteinüri selektiftir, albüminüri mevcuttur. 25 yıl sonra %5’den az olguda kronik böbrek yetmezliği (KBY) gelişir. Erişkinlerde de tedaviye yanıt iyidir ama sık tekrarlar.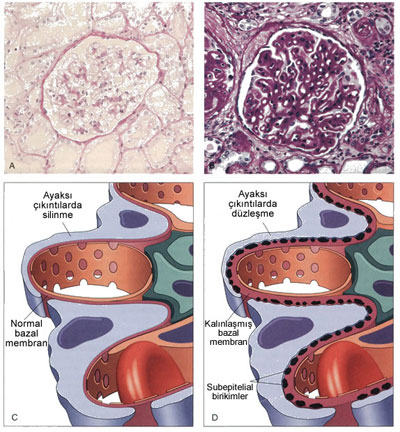 Membranöz Glomerülonefrit
Membranöz Glomerülonefrit
30-50 yaş arası sıktır. Ağır ilerler. Heymann nefritinin insandaki karşılığıdır. Erişkinlerin en sık primer nefrotik sendromu olmakla birlikte olguların %85 ‘i primerdir. %15 ‘i ise bazı sistemik hastalıkların sekonder olarak glomerülleri etkilemesi ile sekonder olarak gelişir.
Sekonder Membranöz GN nedenleri:
1- Enfeksiyon (Kronik hepatit B, sifiliz, Schistosomiasis, malaria)
2- Malign epitelyal tümörler ( Akciğer ve kolon karsinomları, malign melanom)
3- SLE
4- İnorganik tuzlar (Altın, merküri)
5- İlaçlar (Penisilamin, kaptopril)
6- Metabolik hastalıklar (DM, tiroidit)
Primer olgularda çoğunda glomerül antijenlerine yönelik antikorlar reaksiyonu başlatır. Otoimmün olabileceği düşünülmektedir. Sekonder formlarında dolaşan immun kompleksler etkilidir. Nötrofiller görülmediğinden, C5b-9 membran saldırı kompleksinin (litik kompleman) olaylara sebep olduğu ileri sürülmüştür.
- IM ile GBM’de diffüz kalınlaşma görülür ve bu nedenle membranöz GN adı verilmiştir.
- EM ile erken dönemde subepitelyal birikimler, ve bunların birbirinden GBM matriksinin oluşturduğu dikensi çıkıntılar ile ayrıldığı izlenir (diken – kubbe görünümü). Zamanla dikensi çıkıntılar birleşerek birikimleri GBM içine alır. Podositlerin ayaksı çıkıntıları silinir. Zamanla birikimler kaybolup yerinde GBM içinde boşluklar bırakır. Bunlarda GBM benzeri madde ile doludur. Bazal membran kalınlaşır. Zamanla glomerüler skleroze olur ve tamamen hyalenleşir.
- İmmunfloresan ile Ig ve komplemanın (IgG, C3) GBM boyunca granüler birikimi izlenir.
Filtrasyon barierinin iki elemanında defekt vardır. Bazal membran kalınlaşmıştır ve podositlerin ayaksı çıkıntıları silinmiştir. Bu nedenle proteinüri yoğun ve nonselektiftir. Kortikosteroide yanıt kötüdür. Globulin de kaybedilir. % 60 olguda proteinüri kalıcı olup %40 olguda 2-20 yıl sonra KBY gelişir. %10-30 olguda proteinüri kısmen veya tamamen kaybolup iyi gidişlidir.
Fokal segmental Glomerüloskleroz
Glomerüllerin bir kısmında (fokal); her glomerülünde yalnız belirli bir bölgesinde (segmental) bozukluk oluşur. Nefrotik sendromlar içerisinde en kötü prognoza sahiptir.
FSG nedenleri:
1- HIV enfeksiyonu, eroin alışkanlığı, orak hücreli anemi, ağır obesite
2- Diğer glomerülonefritlerde sekonder gelişim (örneğin IgA nefropatisinde)
3- Unilateral agenezis, reflu yada hipertansif nefropatiler de glomerüler ablasyon nefropatisi
4- Primer hastalık olarak (idiopatik FSG)
Glomerüler ablasyon nefropatisi: Herhangi bir nedenle glomerüllerin bir kısmı zedelendiğinde (fokal hastalık), sağlam kalan glomerüllerin aşırı yüklenmeye bağlı zedelenmesidir.Kompansatuar hipertrofi olur ama viseral epitel hücreleri çoğalmaz. Bu nedenle glomerülün yapısı bozulur ve fibrozis gelişir.
Nefrotik sendromlu çocuk ve erişkinlerin %10-35’inde primer FSG vardır. Çocuklarda lipoid nefrozla karışabilir. Klinik farklılıkları: 1)Nefrotik sendrom olduğu halde hematüri, hipertansiyon insidansı yüksektir ve proteinüri seçici değildir 2)Kortikosteroide yanıt kötüdür 3)10 yıl içinde %50 olguda KBY gelişir. 4) Sklerotik bölgede IgM ve C3 birikir.
Erişkinlerde prognoz daha kötüdür. Transplantasyon yapılan hastalarda bazen 24 saat içinde olay tekrarlandığından, viseral epitel hasarına yol açan bir mediatörün dolaşımda bulunduğu ileri sürülmüştür. Bazı olgularda genetik defekt belirlenmiştir. NPHS1 gen ürünü olan ve slit diafram ile ilişkili olan nefrinin yapısında bozulma ile Finnish tipi konjenital nefrotik sendrom oluşur. NPHS2 gen defekti ile, yine slit diafram ile ilişkili olan podosin etkilenir ve çocuklarda steroide dirençli nefrotik sendrom oluşur. Prenatal gen analizi ile tanı konabilir.
İlk olarak genellikle jukstamedüller (medullanın hemen üzerindeki kortekste yerleşen) glomerüller etkilenir. Zamanla tüm korteksde görülebilir.
• Işık mikroskobunda mezenjial matriks artar, bazal membran kollapsı, hyalen birikimi (hyalinozis) ve lipid damlacıkları görülür. Nadiren tüm glomerül skleroze olur (global skleroz).
• İmmünfloresan ile IgM ve kompleman mezenjiumda birikir.
• EM ile visseral epitel ayaksı çıkıntılarını yitirmiş ve GBM’den belirgin olarak ayrılmıştır.
Zamanla total glomerül sklerozu, tübüler atrofi, intersisyel fibrozis gelişir ve diğer kronik glomerülonefritlerden ayrılamaz. Çocuklarda daha iyi prognozludur. Transplantasyondan sonra %25-50 tekrarlar.
Membranoproliferatif Glomerülonefrit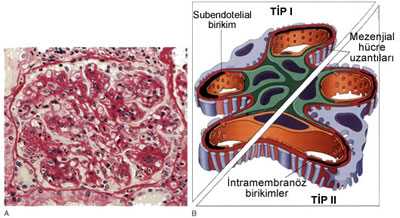
Bazal membran ve mezenjium değişiklikleri, glomerül hücrelerinde proliferasyon ve lökosit infiltrasyonu ile karakterlidir (Mezenjiokapiller GN). Çocuk ve erişkinlerde primer nefrotik sendrom olgularının %5-10’unu oluşturur. Bazen yalnızca hematüri ile nefritik tablo oluşturur. Bazı olgularda nefritik-nefrotik bulgular verir. Primer olgular iki alt gruba ayrılır: Tip I ve Tip II MPGN
- Işık mikroskobu ile her iki tip benzer özellikler gösterir. Glomerüller büyük, mezenjial hücreler ve kapiller endotel hücreleri çoğalmış olup lökosit infiltrasyonu vardır. Lobüler görünürler. GBM kalınlaşmış olup çift kontürlü görülür.
- Olguların 2/3’ü tip I MPGN’ tir. EM ile subodentalyal birikim, İmmunfloresan ile granüler C3 birikimi yanı sıra IgG ve erken kompleman (C1q-C4) komponentleri bulunur. Erken kompleman varlığı, komplemanın klasik yoldan uyarıldığını yani ön planda immünkomplekslere bağlı olarak geliştiğini düşündürür. Tip I MPGN primer olgular dışında Hep B ve C antijenemisi, SLE, enfekte AV shuntlar, sekonder enfeksiyonlarda da immünkomplekslere bağlı olarak gelişebilir.
- Tip II MPGN de lamina densa tabaksı düzensiz, kurdela gibi, elektron yoğun bir yapıya dönüşür ve yoğun birikim hastalığı da denir. C3 birikimi mezenjiumda halka tarzında agregat yapar IgG ve erken kompleman bileşikleri genellikle yoktur. Kompleman alternatif yoldan aktive olur. Hasta serumlarında C3NeF (nefritik faktör) bulunur ve alternatif yol C3 konvertazı bağlar. Konvertaz stabilize olur ve C3 degradasyonu ile hipokomplemanemi yapar. C3NeF otoantikordur ve diğer otoimmun hastalıklarda olduğu gibi genetik yatkınlık sözkonusudur.
Büyük çocuk yada genç erişkinlerde klinik bulgu verir. Bazen hızla ilerleyen GN’e dönüşebilir. Prognoz kötüdür (10 yıl içinde%50 KBY)Tip II MPGN prognozu daha kötü olup, dolaşımdaki nefritik faktör nedeni ile, transplantasyon sonrası tekrarlama eğilimindedir.
Sekonder MPGN
• Kronik immünkompleks hastalıkları: SLE, HBV, HCV, HIV, şistozomiazis, kronik viseral apseler, endokardit, enfekte ventriküloatrial şantlar
• Alfa-1 antitripsin yetmezliği
• KLL ve lenfoma gibi maliğn hastalıklar
• Herediter kompleman düzenleyici protein yetmezlikleri
AKUT NEFRİTİK SENDROM
1. Hematüri
2. Bir miktar oligüri ve azotemi
3. Hipertansiyon
Bir miktar proteinüri ve ödem olsada nefrolit sendromdaki kadar belirgin değildir. Glomerül hücrelerinde proliferasyon ve lökosit infiltrasyonu bulunur. İnflamasyon kapiller duvarı zedeler, eritrositler idrara geçer ve eritrosit slendirleri oluşur.. Hemodinamik değişiklikler ile glomerül filtrasyonu azalır. SLE, mikroskobik poliarterit gibi sistemik hastalıklar veya primer glomerül hastalıkları akut nefritik sendrom yapabilir.
Akut proliferatif Glomerülonefrit
(Poststreptokoksik, Postenfeksiyöz)
Diffüz proliferatif GN en sık görülen glomerül hastalıklardan biridir. İmmun kompleksler ile oluşur. Eksojen antijenle oluşanın prototipi postinfeksiyöz GN’dir. Pnömokok, Stafilokok, kabakulak, kızamık, suçiçeği ve Hep B sonrasında da oluşabilir. Endojen antijenlerle oluşanın prototipi SLE’de görülen lupus nefritidir.
Poststreptokoksik GN
A grubu streptokok enfeksiyonundan 1-4 hafta sonra ortaya çıkar. İlk enfeksiyon faranjit veya cilt enfeksiyonudur. Hipokomplemanemi, GBM’de granüler IgG ve kompleman birikimi tipik bulgulardır.
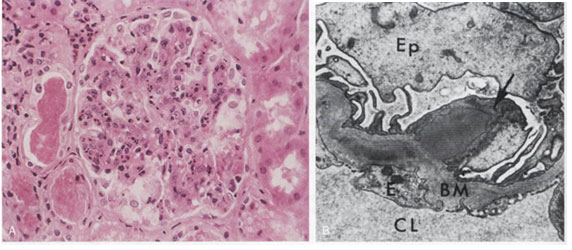
• Işık mikroskobunda tüm glomerüllerde (diffüz) kapiller tomurcuklarda hücresellik artışı, endotel-mezenjial hücrelerde şişme ve prolifasyon; nötrofil ve monosit infiltrasyonu ile ağır olgularda yarımay oluşumu
• EM ile subendotelial, intramembranöz ve daha sık olarak subepitelyal birikimler (hörgüç şeklinde)
• İmmunfloresan ile IgG, IgM ve C3 birikimi… Birikimler 2 ay sonra genellikle kaybolur.
Klinik bulgular ani başlar. Belirgin hematüri, az oranda proteinüri vardır. ASO yüksektir. Oligüri, azotemi ve hipertansiyon hafif-orta derecededir. Çocuklarda %1 olguda hızlı gelişen GN veya böbrek yetmezliği oluşur. Bir kısmı kronik glomerülonefrite ilerlerken, gerisi iyileşir. Erişkinlerde %15-50 olgu kronikleşir.
Hızla İlerleyen (Kresentik) Glomerülonefrit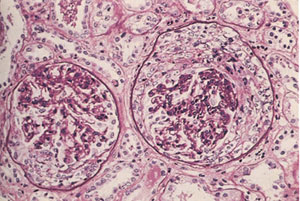
En kötü prognozlu glomerül hastalığıdır. Klinik olarak hızlı ve ilerleyici renal fonksiyon kaybı, ağır oligüri ile tedavi edilmezse, birkaç hafta-ay içerisinde böbrek yetmezliği ile ölüm oluşur. Glomerüllerin çoğunda yarım ay (kresent) varlığı izlenir. Bowman kapsülünde parietal epitel hücrelerinde çoğalma, monosit ve makrofaj infiltrasyonu sonucunda gelişir. Çoğu immünolojik mekanizma ile oluşur.
3 alt grubu vardır:
- Tip I RPGN = Anti - GBM hastalığı olup IgG ve C3’ün GBM’de diffüz birikimi vardır. Antikorlar akciğer alveol bazal membranı ile çapraz reaksiyon verip Goodpasture sendromu oluşturabilir. Antikorlar tip IV kollajenin nonkollajenöz bölgesine (alfa-3 zinciri) yöneliktir.İmmünfloresan ile bazal membran boyunca lineer birikimler görülür. Akciğer kanaması, böbrek yetmezliği birlikte olur. Plazmaferezden faydalanabilirler.
- Tip II RPGN : İmmun kopmleks hastalığıdır. Poststrep GN, SLE, IgA nefropatileri ve Henoch – Schönlein purpurasında komplikasyon olarak gelişebilir. İmmünfloresan ile birikimler granülerdir. Plazmaferez faydasızdır. Altta yatan neden tedavi edilmelidir.
- Tip III RPGN (pausi immun tip)’de immunfloresan veya EM ile antikor ve immun kompleks varlığı belirlenemez. Çoğunda serumda ANCA (antinötrofil sitoplazma antikoru) bulunur. Bazı vaskülitlerde rol oynayan bir antikordur. Tip III RPGN mikroskobik poliarterit ve Wegener granulomatozu ile ilişkili olabilir. Ama çoğu olguda tek başına ve idiopatiktir.
Her üç tipte de olguların çoğu idiopatik olup tüm tiplerde ortak bulgu ağır glomerül hasarıdır.
Böbrekler büyük soluk, olup, korteksde peteşiler mevcuttur. Belirgin yarımay oluşumu vardır. Parietal hücreler çoğalarak Bowman boşluğunu doldurur ve glomerülü sıkıştırır. Fibrin birikimi belirgin olup yarımay oluşumunu bunun başlattığı düşünülmektedir. Subepitelyal birikim görülebilirse de asıl bulgu EM ile GBM’de yırtıklar oluşmasıdır. Zamanla yarımay yapıları skleroze olur. Tedavide steroid ve sitotoksik ilaçlar kullanılır. Yinede olguların çoğunda, dializ ve transplantasyon gerekir.
Ig A Nefropatisi
(BERGER HASTALIĞI)
Çocuk ve genç erişkinlerde ÜSYE takiben bir-iki gün içinde gros hematüri ile ortaya çıkar. Hematüri birkaç gün sürer ve birkaç ayda bir tekrarlar. Hafif proteinüri ve nadiren nefrotik sendrom gelişebilir. Dünyada en sık görülen glomerüler hastalık olup tekrarlayan mikroskopik veya gros hematürinin en sık nedenidir. Mezenjiumda IgA birikir. Kesin tanı immünfloresan inceleme ile konur. Bazı HLA ve kompleman fenotipleri ile ilişkili olduğundan genetik etki sözkonusu olabilir. Virüs, bakteri, gıda proteinleri gibi etkenlerle mukozal IgA sentezinin arttığı ve bunların mezenjiuma yerleşerek alternatif kompleman yolunu uyardığı öne sürülmüştür. Çöliak hastalığında ve IgA komplekslerinin temizlenmesinde görev alan karaciğerin hastalıklarında Berger hastalığı oluşabilir. Henoch-Schönlein purpurasında da benzer şekilde mezenjial IgA birikimi görülür ve iki hastalığın ilişkili olduğu ileri sürülmektedir.
Işık mikroskobisinde bulgular değişkendir:
• Glomerül normal olabilir.
• Mezenjiumda genişleme ve proliferasyon (mezenjioproliferatif GN)
• Bazı glomerüllerde segmental proliferasyon (fokal proliferatif GN)
• Nadiren belirgin kresentik GN
İmmunfloresan ile karakteristik olarak mezenjiumda IgA birikimi; eşlik eden C3 ve az oranda IgG veya IgM birikimi görülür. Erken kompleman bileşikleri genellikle görülmez. Çünkü Ig A komplemanı alternatif yoldan uyarır. EM ile, mezenjiumda birikimler görülür.
Çocuklarda iyi gidişli olmakla birlikte erişkinlerde yavaş olarak ilerler. 20 yıl sonra olguların %15-40’ında KBY gelişir. Transplantasyondan sonra tekrarlayabilir.
HEREDİTER NEFRİTLER
Alport sendromu
Nefrit, sağırlık, değişik göz bozuklukları (lens dilokasyonu, posterior kataraktlar, korneal distrofi). Erkeklerde daha sık ve kötü gidişlidir. 5-20 yaşta belirgin böbrek yetmezliği görülür. X’e bağlı, OR yada OD geçiş gösterebilir.
Segmental glomerüler proliferasyon ve/veya sklerozis; mezenjial matriks artışı vardır. Nötral yağ ve mukopolisakkarit birikimi ile epitel hücrelerinde köpüksü görünüm ile köpük hücreleri oluşur. Zamanla glomeruloskleroz, vasküler daralma, tübüler atrofi ve intestisyel fibrozis gelişir. EM ile bazal membranlarda incelme alanları görülür.
X’e bağlı geçiş gösterenlerde tip IV kollajen yapısında (alfa-5 zinciri) defekt belirlenmiştir.
İnce Bazal Membran hastalığı (Beniğn familial hematüri)
GBM’ de incelme (150- 250 nm), familial asemptomatik hematüri ve iyi prognozla karakterlidir. Hafif-orta derecede proteinüri olsa da, renal fonksiyon bozulmaz. Sağırlık ve göz bozuklukları görülmez.
Fabry hastalığı
Lizozomal depo hastalığıdır. X’e bağlı geçer. Galaktoserebrozid metabolizması bozuk olup birikir. KVS, SSS, RES hücreleri etkilenmekle birlikte en ağır bulgular böbrekte olup hastaların çoğu orta yaşta böbrek yetmezliği ile kaybedilir. Hemodializ ve/veya böbrek transplantasyonu gerekir. Başka bir bulgusu ise kırmızı-mavi cilt nodülleridir (anjiokeratom) .
Kronik Glomerülonefrit
Başlatıcı lezyonun bulguları kaybolmuştur. Fokal glomerüloskleroz, membranöz GN, membranoproliferatif GN gibi glomerüler hastalıkların son dönemidir. %20 olguda hastalık öyküsü yoktur. Genç ve orta yaşlılarda başlangıç sıktır.
Böbrekler simetrik olarak kontrakte olup yüzeyleri kırmızı-kahverenkte ve diffüz olarak granülerdir. Glomerül ve Bowman kapsülünde yaygın skar oluşumu ve sonuçta glomerüllerin tamamen hyalinize olması sözkonusudur. Afferent – efferent artrioller arasında kan akımı gerçekleşemez. İskemiye bağlı belirgin interstisyel fibrozis, tübül atrofisi oluşur. Hipertansiyona sekonder olarak küçük – orta çaplı arter duvarları kalınlaşmış, lümenleri daralmıştır. İntersisyel lenfosit infiltrasyonu görülür. Tüm yapılar zedelendiğinden patolojinin glomerül, tübülointersisyel böge veya damar kökenli olup olmadığı ayrılamaz ve son dönem böbrek hastalığı olarak isimlendirilir. Tedavi yapılmazsa üremi ve ölüm kuraldır. Dializ ve transplantasyon gereklidir.
Dializ Lezyonları:
• Arteriel intimal kalınlaşma
• Fokal kalsifikasyonlar, tübüller içerisinde kalsiyum oksalat kristalleri
• Edinsel kistler
• Adenom ve adenokarsinom sıklığında artış
Üremi Komplikasyonları:
• Üremik perikardit
• Sekonder hiperparatiroidizm sonucu nefrokalsinozis, renal osteodistrofi
• Hipertansiyona bağlı sol ventrikül hipertrofisi
• Üremi sonucu diffüz alveol hasarı ile üremik pnömonitis