LENF NODUNUN YAPISI
Histoloji :
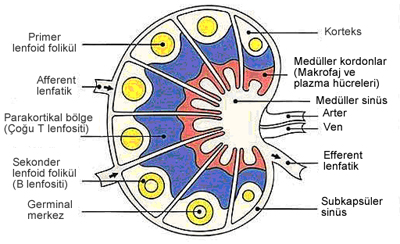
Lenf nodları kapsüllü sferik yapılardır. Konveks kenarı ve konkav kenarı (hilus) bulunur. Hilusda arter ve sinirler içeri girer; venler ve lenfatikler dışarı çıkar. Bağ dokusundan oluşan kapsül içeriye dallar verir. Her lenf bezi dış ve iç korteks ve medulladan oluşur. Dış kortekste en dışta subkapsüler sinüs bulunur. Lenfoid nodüller (folküller) dış korteksde yerleşir. İç korteksde nodüller az olup T lenfositleri baskındır. Medullada B lenfositler ve plazma hücrelerinden zengin kordonlar arsında dilate kapiller benzeri medüller lenfoid sinüsler bulunur. Tüm sinüsler retiküler hücreler ve makrofajlarca döşelidir ve lenf sıvısı içerirler. Büyük dendritik (foliküler) hücreler ise antijen tanıtıcı hücrelerdir. Lenf sinüslerden geçerken antijen ve debrisin çoğu makrofajlarca temizlenir. Enfeksiyon ve antijenik uyarı ile lenf nodları büyürler ve aktif germinal merkezler (sekonder foliküller) oluşur. Plazma hücrelerinin oranı artar.
NEOPLAZİLER
- Lenfoid neoplaziler: Olguların çoğunda neoplazik hücre, lenfosit diferansiasyonunun belirli bir aşamasına benzer. Bu özellik tanı ve sınıflamada kullanılır.
- Myeloid neoplaziler: Myeloid seri hücrelerini (eritroid, granülositik, ve/veya trombositik) oluşturan kök hücreden gelişir. Akut myeloid lösemide immatür progenitör hücreler, kemik iliğinde birikir. Myelodisplastik sendromda inefektif eritropoez ve periferik kanda sitopeni oluşur. Kronik myeloproliferatif hastalıklarda terminal diferansiye bir yada daha fazla myeloid elemanın (ör.granülositler) üretimi artmıştır.
- Histiositozlar nadir görülür. Makrofaj ve dendritik hücrelerin proliferatif lezyonlarıdır. Langerhans hücreli histiositozlar bu grupta yer alır.
Etyoloji ve patogenez
Kromozomal translokasyon ve onkogenler:
B lenfositleri, antijenle uyarıldığında, germinal merkezlerde diferansiye olurlar. Bu sırada immünglobülin genlerinde, çift sarmallı DNA kırıklarınıda içeren modifikasyonlar oluşur. Bu sırada oluşan hatalar onkogenik düzenlemelere ve B hücreli maliğnitelere yol açabilir. T hücre reseptör genleri stabil olduğundan, T hücreli lenfomalar daha az görülür. Myeloid neoplazilerde de kromozomal translokasyonlar sıktır ama mekanizmaları açıklanamamıştır.
Herediter genetik faktörler:
Bloom sendromu, Fankoni anemisi ve ataksi telenjiektazi gibi genomik instabiliteye yol açan hastalıklarda akut lösemi sık görülür. Ayrıca Down sendromu ve tipI Nörofibromatozis, çocukluk çağı lösemileri ile ilişkilidir.
Virüsler:
HTLV-1 :Erişkin T hücreli lösemi ve lenfoması
EBV: Burkitt lenfoma, Hodgkin lenfoma olgularının %30-40 kadarı, immünyetmezlikte görülen B hücreli lenfomalar ve nadir NK hücreli lenfomalar
KSHV/HHV-8: Sıklıkla plevral kavitede maliğn effüzyon şeklinde görülen B hücreli lenfoma
Çevresel etkenler:
Helikobakter pilori enfeksiyonu, gastrik B hücreli lenfoma ile ilişkilidir. Gluten enteropatisi T hücreli intestinal lenfomaya yol açabilir. HIV enfeksiyonunda poliklonalB hücre aktivasyonu ve belirgin germinal hiperplazi nedeni ile B hücreli lenfomalar gelişebilir. Ayrıca hastalarda T hücre yetmezliğine bağlı oluşan EBV enfeksiyonu da, lenfoma riskini arttırır.
İatrojenik faktörler:
Radyoterapi ve bazı kemoterapi ajanları, hematolenfoid progenitör hücrelere mutajenik etki yaparak, myeloid ve lenfoid neoplazi riskini arttırırlar.
LENFOİD NEOPLAZİLER
-
LENFOİD NEOPLAZİLER
Lösemi: yaygın kemik iliği tutulumu ve genellikle eşlik eden periferik kanda çok sayıda tümör hücresi ile karakterlidir. Lenfoma ise dokuda kitle oluşumu ile karakterlidir. Lenfositik lösemi ve lenfoma arasındaki sınır her zaman net değildir. Tedavi edilemeyen lenfomalarda yaygın kemik iliği tutulumu ve periferik kanda tümör hücreleri ile lösemik dönüşüm nadir değildir. Bu nedenle lösemi ve lenfoma terimleri, daha çok, tanı anındaki yayılım düzeyi ile ilişkilidir.
Non-Hodgkin lenfomaların (NHL) 2/3’ ü ve Hodgkin lenfomaların (HL) hemen tümü, genellikle 2 cm çaptan büyük, ağrısız, lokalize yada yaygın lenf nodu büyümesi ile dikkati çeker. Non- Hodgkin lenfomaların 1/3’ ü ise cilt, mide, beyin gibi ekstranodal bölgelerde yerleşir. Lösemi, infiltrasyon nedeni ile kemik iliğinde hematopoezin süpresyonuna bağlı semptomlarla kendini gösterir. Lenfositik lenfomalar karaciğer ve dalağı infiltre ederek büyütür. Plazma hücre tümörleri iskelet sisteminde lokal kemik destrüksiyonu ve patolojik kırıklara bağlı oluşan ağrı ile dikkati çeker.Lenfoid neoplazilerin özellikleri:
- Kesin tanı için lenf nodu yada tutulan bölgelerin patolojik incelemesi gereklidir.
- Neoplazik hücreler monoklonal olup aynı antijen dizilimini gösterir ve aynı proteini üretirler. Reaktif proliferasyonlar ise poliklonaldir.
- Çoğu (%80-85) B hücre kökenlidir. Geri kalanların çoğu T hücreli; nadiren NK hücreli lenfomalardır.
- Çoğu kez immün sistemde normal yapı ve fonksiyon bozulur ve immün anomaliler oluşur.
- Neoplastik B yada T hücreleri, normal hücrelerin davranışlarını taklit eder. Foliküler lenfomalar nodüler yada foliküler büyüme paterni gösterir. T hücreli lenfomalar parakortikal bölgelerde yerleşir.
- Hodgkin lenfoma belirli bir düzeni izleyerek yayılır ve bu nedenle tedavi için evreleme çok önemlidir. NHL ise tanı anında çoğu kez sistemik yayılım gösterir. Bu nedenle evreleme prognoz için önemli olsa da, tedavi için çok önemli değildir.
Prekürsör B- ve T- hücre tümörleriAkut lenfoblastik Lösemi/Lenfoma
Lenfoblast denilen immatür, prekürsör lenfositlerden oluşurlar. Çoğu (%85) prekürsör B hücrelerinden oluşan, yaygın kemik iliği ve değişken periferik kan tutulumu gösteren çocukluk çağı akut lösemileridir. Prekürsör T hücreli olanlar daha az görülüp, çoğu adolesan erkeklerde timik tutulum ile karakterli lenfoma şeklindedir. Morfolojik özellikleri benzediğinden iki grup arasındaki ayırım immünfenotipleme ile yapılır. Klinik özellikleride benzer. Olguların çoğu 15 yaş altında olup beyaz ırkta ve erkek çocuklarında biraz daha sık görülür. pre-B ALL 4 yaş civarında; pre-T ALL adolesan dönemde sıktır. Her iki tümör erişkinlerde görülebilirse de, çocuklara göre nadirdir.
• Morfoloji: Tedavi şekilleri farklı olduğundan ALL ve AML ayırımı önemlidir. Lenfoblastların, myeloblastlardan farklı olarak, kromatini yoğun, nükleolü belirsiz olup dar ve agranüler sitoplazması vardır. Kesin tanı B ve T lenfositlerine spesifik belirleyicilerin antikor kullanarak gösterilmesi ile konur. Lenfoblastların peroksidaz pozitif granülleri yoktur ve sitoplzamada PAS pozitif materyal içerirler. Lenfoma benzeri tablo oluşturan daha çok pre-T tipidir. %50-70 olguda eşlik eden mediastinal kitle (timik tutulım) ve lenfadenopati ile splenomegali belirlenir. Hangi tipi olursa olsun, normal doku yapısını bozan lenfoblast infiltrasyonu, bazı hücrelerde çentikli görünüm, yüksek mitoz oranı, ölen hücreli temizlemek için gelen makrofajlarla oluşan yıldızlı gök manzarası benzeri görünüm mevcuttur.
• İmmünfenotip: pre-B ve pre-T hücrelerinde bulunan TdT, >%95 olguda bulunur. T ve B hücre ayırımı için spesifik belirleyiciler kullanılır. Pre-B hücresi CD19, CD10; pre-T hücresi CD1, CD2, CD5 ve CD7. Erken pre-B ALL hücresinde sitoplazmik IgM bulunmaz. Erken pre-T hücresinde, geç pre-T hücresinden farklı olarak CD3, CD4 ve CD( bulunmaz.
• Sitogenetik ve moleküler genetik: %90 olguda lenfoblastlarda sayısal yada yapısal kromozom değişikliği vardır. En sık hiperploidi (>50 kromozom), daha nadiren poliploidi ve t(12;21), t(9;22) (Filadelfia kromozomu) ve t(4;11) translokasyonları görülür.
• Klinik: Birkaç gün yada hafta içerisinde ani klinik başlangıç; kemik iliği süpresyonuna bağlı bulgular; kemik hassasiyeti ve ağrısı; yaygın lenfadenopati, hepatosplenomegali (AML’ den sık), testis tutulumu; meningeal yayılıma bağlı santral sinir sistemi bulguları (AML’ den sık); pre-T ALL’ de büyük mediastinal damarlar ve solunum yollarına bası bulguları;
• Prognoz: Santral sinir sistemi profilaksisi ile birlikta agresif kemoterapi %90 remisyon ve en az 2/3 olguda iyileşme ile sonuçlanır. Kötü prognoz bulguları: 1) 2 yaş altı (11. kromozomdaki MLL gen translokasyonu sık) 2) adolesan ve erişkin yaş; 3) periferik kanda 100,000 üzerinde blastik hücre;4) Çocukluk çağında %3, erişkinde %25 olguda belirlenen t(9;22) – Filadelfia kromozomu gibi sitogenetik anomaliler.. İyi prognoz bulguları: 2-10 yaş; düşük sayıda hücre, erken pre-B fenotipi, hiperploidi yada t(12;21) WHO Klasifikasyonu
-
WHO Klasifikasyonu
1) Prekürsör B hücre tümörleri
4) Periferik T ve NK hücre tümörleri
Prekürsör B lenfoblastik lösemi/lenfoma
T hücre prolenfositik lösemi
Büyük granüler lenfositik lösemi
Mikozis fungoides/Sezary sendromu
Periferal T hücreli lenfoma, nonspesifik
Anaplastik büyük hücreli lenfoma
Anjioimmünoblastik T hücreli lenfoma
Enteropati ile ilişkili T hücreli lenfoma
Pannikülit benzeri T hücreli lenfoma
Hepatosplenik T hücreli lenfoma
Erişkin T hücreli lösemi/lenfoma
NK/T hücreli lenfoma, nazal tip
NK hücreli lösemi
2) Periferik B hücre tümörleri
5) Hodgkin Lenfoma
Kronik lenfositik lösemi/ küçük lenfositik lenfoma
B hücre prolenfositik lösemi
Lenfoplazmositik lenfoma
Splenik ve nodal marjinal zon lenfoma
Ekstranodal marjinal zon lenfoma
Mantle hücreli lenfoma
Foliküler lenfoma
Marjinal zon lenfoma
Hairy cell lösemi
Plazmositom/plazma hücreli myelom
Diffüz büyük B hücreli lenfoma
Burkitt lenfoma
Klasik tipler
· Nodüler skleroz
· Karışık hücreli
· Lenfositten zengin
· Lenfositten fakir
Lenfosit baskın
3) Prekürsör T hücre tümörleri
Prekürsör T lenfoblastik lösemi/ lenfoma
Periferik B Hücre Tümörleri
Kronik lenfositik lösemi (CLL)/ Küçük lenfositik lenfoma (SLL)
Bu iki hastalık morfolojik, fenotipik ve genotipik olarak birbirinden ayrılamaz. Aralarındaki tek farklılık, periferik kandaki lenfositoz derecesidir. Çoğu hastada lenfosit sayımı >4000/mm3 olup lösemi tanısı alır. Batılı ülkelerde en sık görülen erişkin çağ lösemisidir. SLL ise NHL olgularının %4 kadarını oluşturur. Her iki tümörde Japonya ve Asya’ da nadirdir.- Morfoloji: Lenf nodunu diffüz infilte eden yuvarlak, hafif düzensiz nüveli, dar sitoplazmali küçük lenfositler görülür. Arada daha büyük prolenfositler ve bunların oluşturduğu, patognomik olan proliferasyon merkezleri vardır. Lösemik formunda kanda bulunan lenfositler frajil olup yayma hazırlanırken zedelenir ve ezilmiş hücreleri oluştururlar. Kemik iliği tutulumu lösemilerin hepsinde, lenfoma olgularının çoğunda belirlenir. Dalakda beyaz ve kırmızı pulpa ile karaciğerde portal traktüs infiltrasyonu görülür.
- İmmünfenotip: CD19, CD20, CD23 ve CD5düşük düzeyde yüzey immünglobülin ekspresyonu (IgM/ IgM ve IgD)
- Kromozom anomalisi ve moleküler genetik: Kromozomal translokasyon nadirdir.
- Klinik: Çoğu 50 yaş üzeri (median yaş 60) erkek hastalardır (M/F: 2/1). Çoğu asemptomatiktir. Kolay yorulma, kilo kaybı, iştahsızlık görülebilir. Jeneralize lenfadenopati, hepatosplenomegali %50-60 olguda mevcuttur. Kemik iliği tutulumu olan SLL olgularında lökopeni; ağır CLL olgularında >200,000/mm3 üzeri periferik lökositoz olabilir. Hipogamaglobülinemi sıktır. %10-15 olguda ise kan elemanlarına yönelik antikorlara bağlı otoimmün hemolitik anemi yada trombositopeni görülür. Ortalama sağkalım 4-6 yıl olsa da, hafif olgular 10 yıl yada daha fazla yaşar. 11q ve 17p delesyonları kötü prognozludur. %15-30 hastada prolenfositik transformasyon yada %10 olguda diffüz büyük B hücreli lenfomaya transformasyon (Richter sendromu) prognozu bozar ve hastalar 1 yıl içerisinde kaybedilir.
Foliküler Lenfoma
ABD’ de en sık görülen NHL olup erişkin lenfomalarının %45’ ini oluşturur. En sık orta yaşta; kadın ve erkekde aynı oranda görülür. Avrupa ve Asya’ da daha nadirdir. Neoplastik hücreler normal germinal merkez B hücrelerine benzer.- Morfoloji: Lenf nodunda nodüler yada nodüler/diffüz patern görülür. değişik oranlarda başlıca iki hücre tipi görülür: 1) sentrosit denilen, düzensiz yada çentikli nüveli, dar sitoplazmalı küçük hücreler (küçük çentikli hücreler) 2) birkaç nükleol ve daha geniş sitoplazma içeren sentroblastlar.. Çoğunda küçük çentikli hücreler baskındır. %10 olguda hafif derecede lenfositoz (<20,000/mm3); %85 olguda kemik iliği tutulumu vardır. Splenik beyaz pulpa ve karaciğerde portal infiltrasyon sıktır.
- İmmünfenotip: CD19, CD20,CD10, yüzey immünglobülinleri belirlenir. CD5 (-).>%90 BCL2 protein ekspresyonu (normal foliküler hücrelerinden farkı); hemen tümünde BCL6 ekspresyonu
- Sitogenetik ve moleküler genetik: %90 olguda t(14;18) sonucu IgH lokusu ile 18. kromozomdaki BCL2 lokusu birleşir ve BCL2 ekspresyonu artar. Apoptoz engellenir ve neoplastik B hücreleri çoğalır.
- Klinik: Ağrısız, jeneralize lenfadenopati ile bulgu verir. Ortalama sağkalım 7-9 yıldır. Tedaviye yanıt vermez. %30-50 olguda diffüz büyük B hücrei lenfomaya traansforme olur. Prognoz bozulur.
Diffüz Büyük B Hücreli Lenfoma
Tüm NHL olgularının %20; agresif lenfomaların %60-70 kadarını oluşturur. En sık 60 yaş civarında ve erkeklerde biraz daha sık görülür. Çocukluk çağı lenfomalarının %5 kadarını oluşturur.- Morfoloji: Nispeten büyük boyutlu ( küçük lenfositten 4-5 kat büyük) lenfositler ve diffüz büyüme paterni gösterir. Çoğu kez veziküler nüveli, bazen multilobe yada çentikli nüveli hücreler görülür. Nükleol birkaç adet olup periferde yerleşebileceği gibi tek santral nükleol görülebilir. Hücreler soluk yada bazofilik geniş sitoplazmalıdır. Anaplastik tiplerinde multinükleer hücreler (Reed-Sternberg benzeri) bulunabilir.
- İmmünfenotip: CD19, CD20, bazılarında CD10 ve BCL6, çoğunda yüzey Ig var. Hepsi TdT (-).
- Sitogenetik ve moleküler profil: BCL6 mutasyonları, c-MYC muatsyonları, %10-20 olguda t(14;18) (bunlarda BCL6 mutasyonu yok)
- Onkogenik virüsler ile ilişkili subtipler: Ağır T hücre yetmezliğinde (ileri evre HIV enfeksiyonu, ağır kombine immün yetmezlik, transplantasyon) EBV etkisi ile gelişir. Ayrıca ileri evre HIV enfeksiyonunda ve nadiren HIV negatif yaşlılarda görülen vücut kavite lenfomalarında tümör hücreleri KSHV/HHV8 ile enfektedir.
- Klinik ve prognoz: Tek bir nodal yada ekstranodal bölgede, hızla büyüyen semptomatik kitle ile karakterlidir. Waldeyer halkası, orofaringeal lenfoid doku sıklıkla tutulur. Karaciğer ve dalakta primer yada sekonder tutulum sonucu büyük kitleler oluşur. Kemik iliği geç dönemde tutulur ve lösemik tablo nadirdir. Tedavi edilmez ise öldürücüdür. Yoğun kombine kemoterapi ile %60-80 komplet remisyon ve yaklaşık %50 olguda iyileşme mümkündür.
Burkitt Lenfoma
1) Afrika tipi (endemik), 2) sporadik; 3) HIV enfeksiyonunda görülen agresif lenfomaların bir alt grubu olmak üzere farklı tipleri vardır. Tümünde histolojik görünüm aynı olsada bazı klinik, genotipik ve virolojik farklılıklar vardır.- Morfoloji: Orta boyutlu, birkaç nükleolü olan nüveler içeren, bazofilik yada amfofilik sitoplazmalı lenfoid hücrelerden oluşan infiltrasyon görülür. Mitoz ve apoptoz sıktır. Kalıntıları fagosite eden makrofajlar nedeni ile yıldızlı gök manzarası oluşur. Kemik iliği tutulumu varsa, 2-5 nükleollü, mavi sitoplazmalı, berrak sitoplazmik vakuolleri olan hücreler şeklinde görülür.
- İmmünfenotip: Yüzey IgM, monotipik hafif zincirler, CD19, CD20, CD10 ve BCL6 gibi matür B hücre belirleyicileri
- Sitogenetik ve moleküler genetik: Tümünde 8. kromozom da yerleşen c-MYC geninde translokasyon vardır. Çoğu kez diğer bölge IgH loküsüdür t(8;14). Endemik tümörlerin tümünde; AIDS hastalarının %25’i, sporadik olguların %15-20’ sinde latent EBV enfeksiyonu vardır.
- Klinik: Endemik ve sporadik olguların çoğu çocuk ve genç erişkinlerde görülür. ABD’ de çocukluk çağı lenfomalarının %30’ u Burkitt lenfomadır. Çoğu ekstranodal yerleşir. Endemik olanlar mandibula yanı sıra böbrek, over ve adrenal gland gibi abdominal organlarda yerleşir. Sporadik olgular daha sık olarak ileoçekal bölge ve peritonda yerleşir. Kemik iliği ve periferik kan tutulumu nadirdir. Çok agresif olmakla birlikte, kısa süreli, yüksek doz kemoterapiye iyi yanıt verir. Çoğu çocuk ve genç erişkin iyileşirken, yaşlılarda kötü davranış gösterir.
Plazma Hücre Tümörleri
Tek bir homojen immünglobülin yada fragmanlarını üretip salgılayan B hücrelerinin klonal proliferasyonudur. Bu proliferasyonlar (diskraziler) çoğu kez maliğn davranış gösterir. Beyaz hücre (lökosit) neoplazilerine bağlı ölümlerin yaklaşık %15’ ini oluştururlar. Kanda belirlenen monoklonal immünglobüline M komponenti denir. Normal plazma hücreleri hafif (L) ve ağır (H) zincir çiftleri üretirken neoplazik olanlar komplet immünglobülinler yanı sıra aşırı miktarda L yada H zincirleri üretirler. Serbest hafif zincirlerine Bence Jones proteinleri denir. Küçük boyutlu olduklarından idrarla kolaylıkla atılırlar ve böbrek yetmezliği yoksa yada çok yoğun değilse, kanda belirlenemezler. Monoklonal gamopati, disparaproteinemi gibi isimler verilen bu grup tümörlerde yer alan tablolar şunlardır:- Multiple myelom (plazma hücre myelomu) En önemli ve en sık semptomatik monoklonal gamopatidir. İskelet sisteminde yaygın odaklar yapar. Nadir görülen soliter myelom yada plazmositom ise kemik yada bazı yumuşak dokularda tek (soliter) kitle yapar.
- Waldenström makroglobülinemisi: IgM düzeyi yüksekiliğine bağlı gelişen hipervizkozite vardır. Lenfoplazmositik lenfoması olan erişkinlerde görülür.
- Ağır zincir hastalığı: Serbest H zincirleri üretip salgılayan bir gruptur. CLL/SLL, lenfoplazmositik lenfoma ve ince barsakta nadir görülen bir tümöral proliferasyon (Akdeniz lenfoması) bu tabloya yol açabilir.
- Primer yada immünosit ile ilişkili amiloidoz: Serbest L zincirleri üreten plazma hücre proliferasyonu olup amiloidoz ile sonuçlanır. Bazı hastalarda belirgin multiple myelom; bazılarında ise yalnızca kemik iliğinde minör klonal plazma hücre proliferasyonu belirlenir.
- Önemi belirlenemeyen monoklonal gamopati: Klinik bulgusu olamayan hastalarda kanda m komponentlerinin belirlenmesidir. Yaşlılarda oldukça sık olmakla birlikte semptomatik olgulara (en sık multiple myelom) dönüşüm nadirdir.
Multiple Myelom
-
Multiple Myelom
Kemik tutulumu baskın olmakla birlikte lenf nodu, cilt gibi diğer bölgelere de yayılabilir. Erkeklerde, yaşlılarda ve Afrika kökenli olanlarda daha sık görülür.- Etyoloji ve patogenez: Myelom hücrelerinin proliferasyonu ve yaşaması, IL-6 başta olmak üzere, bazı sitokinlere bağlıdır. Neoplastik plazma hücreleri ve kemik iliğindeki normal stromal hücreler IL-6 üretir. Serumda IL-6 yüksekliği, kötü prognoz bulgusudur. Plazma hücrelerinin ürettiği MIP1alfa ve NF-kappaB ligand reseptör aktivatörü (RANKL) osteoklastları aktive ederek kemik yıkımına yol açar. 13q delesyonu ve Ig ağır zincir (IgH) bölgesini içeren (14q32) translokasyonlar sıktır.
- Morfoloji: İskelet sisteminde plazma hücrelerinden oluşan multifokal, destrüktif tümör odaklarıdır. Sıklık sırasına göre vertebra, kostalar, kafa tası, pelvis, femur, klavikula ve skapula tutulur. Medüller kaviteden başlayarak süngerimsi kemiği ve sonra kortikal kemiği zedeleyerek patolojik kırıklara yol açar. Radyolojik olarak 1-4 cm çapında, zımba ile delinmiş görünümde lezyonlardır. Nadiren diffüz osteopeni yapar. Kemik iliği hücrelerinin %30’ undan fazlası plazma hücrelerinden oluşur. Normal plazma hücreleri gibi perinükleer berraklaşma (Golgi aparatı) ve eksantrik nüve içerirler. Normal görünümde hücreler, plazmablastlar yada anormal görünümde multinükleer hücreler bulunabilir. Düzensiz immünglobülin üretimine bağlı hücre içi birikimler değişik görünümlere yol açar: alev kırmızısı sitoplazma (alev hücreleri); mavi görünümde üzüm tanesi benzeri sitoplazmik damlacıklar (Mott hücreleri); sitoplazmik Russel cisimcikleri; intranükleer Dutcher cisimcikleri; fibril, kristal ve globüller….Hastalık ilerlediğinde dalak, karaciğer, böbrek, akciğer, lenf nodları ve diğer yumuşak dokulara yayılabilir. Yüksek düzeyde M proteini nedeni ile eritrositler birbirine yapışarak, periferik yaymalarda rulo formasyonuna tol açar. Spesifik olmayıp immünglobülinlerin arttığı SLE ve erken HIV enfeksiyonunda da görülebilir. Hücrelerin periferik kana geçip plazma hücreli lösemi oluşturması nadirdir. Bence Jones proteinlerine bağlı myelom böbreği gelişimi önemli bulgulardan biridir.
- Klinik: 50-60 yaş arası pik yapar. Kemik kırıkları, kronik ağrı ve hiperkalsemiye bağlı komplikasyonlar oluşur. Poliüri böbrek yetmezliğini işaret eder. Normal immünglobülin sentezinin azalması tekrarlayan enfeksiyonlara yol açar. Hücresel immünite genellikle etkilenmez. Enfeksiyonlardan sonra, en sık ölüm sebebi böbrek yetmezliğidir. Bence Jones proteinürisi, tübül epiteline toksik etki yapar. Bazı hastalarda amiloidoz gelişir. Hastaların %99’ unda kanda immünglobülin artışı ve/veya idrarda Bence Jones (hafif zincirler) proteinürisi belirlenir. Serumda en sık (%55) IgG, daha nadiren IgA yapısında M proteinleri bulunur. IgA ve IgG3 salgılayan tümörlerde daha sık olmakla birlikte, %7 olguda hipervizkozite mevcuttur. %1 olgu nonsekretuar olup serum ve idrarda M proteini olmaması, myelomu kesin olarak ekarte ettirmez. Kesin tanı kemik iliği incelemesi ile konulur. Prognoz kötüdür. Tedavi edilmeyen yaygın olgular, 6-12 ay içerisinde kaybedilir. Alkile edici ajan kemoterapisi %50-70 remisyon sağlasa da, ortalama sağkalım 3 yıl civarındadır. Bifosfonatlar kemik yıkımını azaltır. Proteazom inhibitörü olan ilaçlara duyarlıdır. 50 yaş altı hastaların az bir kısmında, allojenik kemik iliği transplantasyonu uzun süreli remisyon sağlayabilir.
Soliter myelom
Kemik yada yumuşak dokuda tek odak yapar. Nadir görülür. kemik lezyonları multiple myelomla aynı odaklarda yerleşirken, yumuşak doku lezyonları akciğer, oronazofarinks veya sinüslerde yerleşebilir. Kemik lezyonları 10-20 yıl içerisinde multiple myeloma dönüşebilir. Yumuşak dokuda yerleşenler lokal rezeksiyonla iyileşebilir.Önemi Belirlenemeyen Monoklonal Gamopati
50 yaş üzerinde sağlıklı hastaların %1; 70 yaş üzerinde %3 kadarında serumda asemptomatik M proteinleri belirlenebilir. En sık monoklonal gamopati nedenidir. 30 yıl içerisinde multiple myeloma ilerleyebilir. Serumda monoklonal protein düzeyi <3 gr/dl olup Bence Jones proteinürisi yoktur.Lenfoplazmasitik lenfoma
Yaşlılarda (6-7. dekat) görülen, B hücre tümörüdür. Neoplastik plazma hücreleri monoklonal M proteini üreterek Waldenström makroglobülinemisi denilen hipervizkozite sendromuna yol açabilir. Multiple myelomdan farklı olarak hafif ve ağır zincir üretimi dengelidir. Bu nedenle hafif zincirlere bağlı amiloidoz yada böbrek yetmezliği görülmez. Kemik lezyonu yoktur. Tanı anında kemik iliği yanı sıra lenf nodu, dalak ve karaciğer tutulumu sıktır. Sinir kökleri, meninksler ve nadiren beyine yayılabilir. Görme bozukluğu, nörolojik bulgular, kanama ve kriyoglobülinemi görülebilir. Ortalama sağkalım 4 yıldır.Mantle Hücreli Lenfoma
4.-5. dekatta ve erkeklerde daha sık görülür. Germinal merkezleri çevreleyen normal B lenfositlerine benzeyen hücrelerden oluşur. Yuvarlak yada çentikli hücreli, küçük lenfositlerdir. Çoğunda jeneralize lenfadenopati, %20-40 olguda periferik kan tutulumu vardır. Kemik iliği, dalak beyaz pulpası, karaciğerde periportal bölgeler ve barsak tutulumu (lenfomatoid polipozis) görülebilir. İmmünfenotip olarak CD19, CD20, CD5, yüksek düzeyde yüzey Ig ağır zincirleri, silkin D1 proteini içerirler. 11. kromozomdaki siklin D1 loküsü ve 14. kromozomdaki IgH loküsünü içeren translokasyon (t(11;14) ile ilişkilidir. Siklin D1 ekspresyonu artarak, hücre siklusunda G1-S fazı geçişini kolaylaştırır. Ağrısız lenfadenopati, %50 splenomegali ve gastrointestinal sistem tutulumu görülebilir. Kötü prognozlu olup ortalama sağkalım 3-4 yıldır. Tedaviye yanıt vermez.Marjinal Zon Lenfomalar
Lenf nodu, dalak yada ekstranodal yerleşimli B hücre tümörleridir. Mukozal bölgelerde görüldüğünde maltoma da denir. Postgerminal bellek B hücrelerinin özelliklerini taşır. Ekstranodal olanlar enfeksiyoz yada otoimmün lezyonlar ile ilişkilidir (Sjögren sendromu, hashimoto tiroiditi, H. Pylori gastriti). Uzun süre lokalize kalırlar ve etken uzaklaştırıldığında gerileyebilirler. Geç dönemde uzak yayılım yada diffüz büyük hücreli lenfomaya transformasyon görülebilir.Hairy Cell Lösemi
Tüm lösemilerin %2’ sini oluşturur. Orta yaşlılarda sıktır. Erkeklerde 4 kat fazla görülür. Hücrelerin saç benzeri çıkıntıları faz-kontrast mikroskobunda daha iyi görülür. Hücrelerin açık mavi sitoplazmaları ve köpük benzeri çıkıntıları vardır. Retikülin fibrilleri ile döşeli ekstraselüler matriks içine yerleştikleri için, kemik iliği aspiratlarına gelmezler. Dalakda kırmızı pulpada infiltrasyon ve beyaz pulpada obliterasyon ile bifteğe benzer kırmızı görünüm olur. Hepatik portal infiltrasyon sıktır. İmmünfenotipik olarak CD19, CD20, CD11c, CD25, CD103, yüzey immünglobülinleri belirlenir. Bellek B hücre özellikleri taşır. En sık masif splenomegali, daha nadiren hepatomegali ve çok az olguda lenfadenopati vardır. Pansitopeni yarısından fazlasında görülür ve enfeksiyonlara yol açabilir. Monositopeniye bağlı atipik mikobakteri enfeksiyonları riski artmıştır. Bazı kemoterapötiklere duyarlı olup uzun süreli remisyon sağlanabilir.Periferik T ve NK Hücre Tümörleri
Periferik T Hücreli Lenfoma, nonspesifik
Küçük, orta ve büyük boyutlu maliğn T lenfositlerinden oluşan diffüz infiltrasyondur. Eozinofil ve makrofajlardan oluşan reaktif hücrelerde infiltrasyona eşlik eder. Sıklıkla belirgin anjiogenez görülür. İmmünfenotipleme ile tanı konur. CD2, CD5, CD3 ve T hücre reseptörleri belirlenir. Bazen CD4 yada CD8 mevcuttur.Jeneralize lenfadenopati, eozinofili, kaşıntı, ateş ve kilo kaybı görülür. Diffüz büyük B hücreli lenfomaya göre daha kötü prognozludur.Anaplastik Büyük Hücreli Lenfoma
2p23 bölgesindeki ALK geninde mutasyon vardır. At nalı benzeri nüve ve geniş sitoplazmalı anaplastik hücreler görülür. Metastatik karsinom ile karışabilir. ALK mutasyonu içeren formlar çocuk ve gençlerde görülür. Yumuşak doku tutulumu sıktır ve iyi prognozludur. ALK negatif olanlar yaşlılarda görülür ve kötü prognozludur.Erişkin T Hücreli Lösemi/Lenfoma
CD4+ T lenfositlerinin tümörü olup HTLV-1 virüsü ile ilişkilidir. Japonya, batı Afrika ve Karaib adaları gibi, virüsün endemik olarak bulunduğu yerlerde daha sık görülür. Cilt lezyonları, jeneralize lenfadenopati, hepatosplenomegali, periferik lenfositoz ve hiperkalsemi ile karakterlidir. Tutulan bölgeler ve periferik kanda multilobe nüveli (çiçek hücreleri) hücreler görülür. Birkaç ay- bir yıl içerisinde, agresif kemoterapiye rağmen, hastalar kaybedilir. HTLV-1 virüsü, ayrıca santral sinir sisteminde progresif, demyelinizan hastalığa yol açabilir.Mikozis Fungoides/Sezary Sendromu
CD4+ T lenfositlerinin maliğn tümörüdür. Cilt tutulumu ön plandadır. İnflamatuar premikotik faz, plak fazı ve en sonunda tümör fazı gelişir. Epidermis ve üst dermisi infiltre eden maliğn hücrelerin serebriform nüveleri vardır. Ekstrakutanöz yayılım lenf nodu ve kemik iliğine olur. Sezary sendromunda jeneralize eksfoliatif eritrodermi ve eşlik eden Sezary (serebriform) hücrelerini içeren lösemi vardır. Yavaş gidişli olup ortalama sağkalım 8-9 yıldır.Büyük Granüler Lenfositik Lösemi
Nadirdir. T hücresi yada NK hücresinden oluşan iki farklı tipi vardır. Erişkinde görülür. Hafif-orta dereceli lenfositoz mevcut olup lenfadenopati ve hepatomegali genellikle görülmez. Mavi sitoplazmalı, azurofilik granüllü hücreler görülür. Nötropeni, anemi sıktır. Romatolojik hastalık sıklığı artar. Felty sendromunda, bazı olgularda romatoid artrit, splenomegali ve nötropeninin altında yatan sebep, bu lösemi olabilir. T hücreliler daha yavaş gidişli olup NK hücreliler kötü prognozludur.Ekstranodal NK/T Hücreli Lenfoma
Asya’ da daha sıktır. Eskiden lethal midline granülom adı verilirdi. En sık nazofarinkste, destrüktif orta hat kitlesi şeklinde, daha nadiren cilt ve testisde yerleşir. Tümör hücreleri küçük damarları infiltre ederek nekrozlara yol açar. Hücre sitoplazmalarında, NK hücreleri gibi azurofilik granüller vardır. EBV ile ilişkili olabilir. cKIT, p73 ve p53 mutasyonları belirlenebilir. HODGKİN LENFOMA
-
HODGKİN LENFOMA
Primer olarak lenfoid dokuları tutan bir hastalıktır. Tek bir lenf nodu veya nod zincirinde gelişip ilişkili olduğu lenf nodlarına yayılır.
- Reed-Sternberg (RS) denilen neoplastik dev hücreler
- Ateş gibi belirgin klinik bulgular ile NHL’dan ayrılır
Reed- Sternberg hücreleri germinal merkez yada post-germinal merkez B hücrelerinden köken alırlar. Bu nedenle Hodgkin lenfoma B hücre tümörü olarak kabul edilmektedir. Genç erişkinlerde en sık görülen maliğn tümörlerden biri olup tanı anında ortalama yaş 32 dir. Tedavide gelişmeler sonucunda çoğu olguda iyileşme sağlanmaktadır.
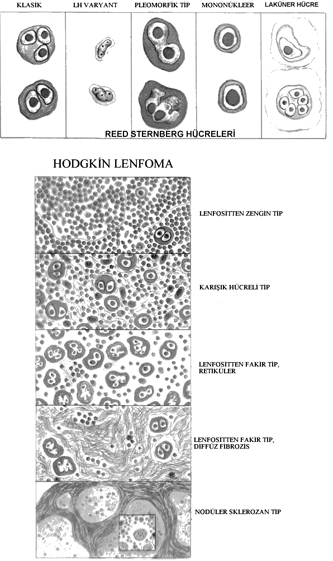
WHO sınıflaması:
- Nodüler sklerozis
- Karışık hücreli
- Lenfositten zengin
- Lenfositten fakir
- Lenfosit baskın
İlk dört grupta Reed-Sternberg hücreleri benzer immünofenotipe sahip iken, lenfosit baskın tipinde farklı yapıda B hücre fenotipi vardır. Bu nedenle ikl dört tipi klasik subtipler olarak kabul edilir.
Morfoloji: Tanı için önemli olan RS hücrelerinin görülmesidir. Çok sayıda loba sahip tek yada birkaç nüveli hücreler olup küçük lenfosit boyutunda, inklüzyona benzeyen nükleolus içerirler. Sitoplazma geniştir. Mononükleer varyantları nükleolusu belirgin, tek nüve içerir. Laküner hücreler nodüler sklerozis tipinde belirgin olup berrak sitoplazma nedeni iel boşluk içinde gibi görülür. Klasik olgularda, Reed-Sternberg hücreleri ölüp piknotik büzüşmüş hücrelere dönüşebilir (mumyalaşma). Lenfohistiositik varyantlar (L&H) polipoid nüveleri ile patlamış mısıra benzer (popkorn). Nükleoller belirgin değildir ve lenfosit baskın tipte görülürler. Enfeksiyöz mononükleoz, solid doku tümörleri ve NHL olgularında Reed-Sternberg hücrelerine benzeyen hücreler görülebilir. Bu nedenle Hodgkin lenfoma tanısı için eşlik eden reaktif lenfosit, plazma hücreleri ve eozinofillerinde görülmesi gerekir. Önce nodal, sonra splenik, sonra hepatik ve en sonunda kemik iliği ve ekstranodal tutulum gösterir. Sınırlı tutulum olan olgularda lokal radyoterapi ile iyileşme sağlanabilir. Evreleme tedavi için önemlidir.- Nodüler sklerozis tipi: En sık görülen tip (%65-70). Laküner hücreler ve lenf nodunu nodüllere ayıran kollajen bantlar görülür. F=M. Alt servikal, supraklaviküler ve mediastinal lenf nodu tutulumu sıktır. Ergenlik ve genç erişkinlerde sık olup nadiren EBV ile ilişkilidir. Prognozu mükemmeldir.
- Karışık hücreli tip: %20-25 olguyu oluşturur. Küçük T lenfositi, eozinofil, plazma hücresi ve beniğn makrofajlardan oluşan hücrelerin arasında diagnostik ve mononükleer Reed-Sternberg hücreleri görülür. Erkeklerde daha sıktır. %70 kadar olguda EBV ile ilişkilidir. İleri yaşta, sistemik semptomlarla birlikte olup sıklıkla ileri evrededir. Yine de prognoz çok iyidir.
- Lenfositten zengin tip: Nadirdir. Mononükleer ve diagnostik Reed-sternberg hücrelerinin sık olması ile lenfosit baskın tipten ayrılır. Bu hücreler CD20 (-), CD15 ve CD30 (+)%40 olguda EBV ile ilişkilidir. Prognozu çok iyi yada mükemmeldir.
- Lenfositten fakir tip: En az görülen tiptir. Lenfositler az olup Reed-Sternberg hücreleri ve pleomorfik tipleri sıktır. Büyük hücreli NHL ile karışır. Yaşlılarda daha sık görülür. HIV (+) kişilerde, geri kalmış toplumlarda daha sık olup sıklıkla EBV ile ilişkilidir. İleri evre ve sistemik bulgular sık olup diğer tiplere göre kötü prognozludur.
- Lenfosit baskın tip: Nadirdir. Olguların %5 kadarını oluşturur. Beniğn histiositler ile karışık küçük lenfosit infiltrasyonu görülür. Tipik Reed- Sternberg hücreleri azdır. Lenfohistiositik varyant (popkorn hücre) sıktır. Eozinofil, plazma hücresi ve nötrofiller az olup nekroz ve fibrozis izlenmez. L&H varyant CD20 gibi B hücre ve BCL6 gibi germinal merkez fenotipi gösteir. %3-6 olguda diffüz B hücreli lenfomaya dönüşür. EBV ile ilişkili değildir. Hastaların çoğu 35 yaş altı, erkeklerdir. Servikal yada aksiller lenfadenopati sık olup mediasten yada kemik iliği tutulumu nadirdir. Tekrarlama riski olsa da, prognoz mükemmeldir.
Etyoloji ve patogenez:
Olguların bir kısmında EBV ile ilişki gösterilmiştir. Reed-Sternberg hücrelerinin salgıladığı IL-5, IL-6, IL-13, TNF, GM-CSF gibi sitokinler reaktif hücrelerin birkimine yol açar. Reed-Sternberg hücreleri anaploid olup klonal kromozom anomalileri içerirler.Klinik: Lenf nodlarında ağrısız büyüme olur. Kesin tanı biopsi incelemesi ile konur. İyi prognozlu tiplerde, hasta genç ise, genellikle evre I yada II ‘de tanı konur. İleri evrelerde yada karışık hücreli ve lenfositten fakir tipte sistemik bulgular mevcuttur. Nadir görülen bir paraneoplastik semptom, alkol alan hastalarda lenf nodu ağrısıdır. Hücresel immünite bozukluğuna bağlı kutanöz anerji sıktır. Tümör tipinden çok, evre önemlidir. Evre I ve IIA’ da iyileşme oranı %90 olup ileri evrelerde bile 5 yıllık remisyon %60-70 civarındadır. Uygulanan uzun süreli kemoterapi ve radyoterapi sekonder kanserlere yol açabilir. Myelodisplastik sendrom, akut myeloid lösemi ve akciğer kanseri en sık görülen tümörlerdir. Ergenlik çeğında, göğüs bölgesine radyoterapi uygulananlarda meme karsinomu görülebilir. Radyoterapinin nonneoplastik komplikasyonları pulmoner fibrozis ve aterosklerozdur. Yeni tedavi yöntemleri ile bu komplikasyonlar azalmıştır.
MYELOİD NEOPLAZİLER
- Akut myeloid neoplaziler: İmmatür myeloid hücrelerin kemik iliğinde birikip normal hematopoezi baskılamasıdır.
- Myelodisplastik sendromlar inefektif eritropoez ve sitopenilerdir.
- Kronik myeloproliferatif bozukluklar termianl diferansiye myeloid hücrelerin üretiminin artışıdır.
Akut Myeloid Lösemi
15-39 yaş arası erişkinlerde daha sık olup nadiren çocuk yada yaşlılarda görülebilir. Çoğunda terminal myeloid diferansiasyonu engelleyen genetik değişiklik vardır. Bu nedenle normal hücrelerin yerinde blastlar birikir. Anemi, nötropeni ve trombositopeni gelişir. Sitotoksik ilaçlar ile lösemik klonun temizlenmesi ve normal hematopoezin sağlanmasına çalışılır. Akut promyelositik lösemide retinoik asidin farmakolojik dozları kullanılır.AML FAB SINIFLAMASI
- MO Minimal diferansiye AML: %2-3. Blastlar myeloblast belirleyicilerini içermez (myeloperoksidaz negatif) ama myeloid seri antijenleri vardır ve ultrastrüktürel olaral myeloblastlara benzerler
- M1 Diferansiasyon göstermeyen AML: %20. İmmatürite belirgin ama >%3 blast peroksidaz pozitif; nadir granül yada Auer çubukçuğu
- M2 Matür AML: %30-40. Granülosit yönünda tam matürasyon; çoğu olguda Auer çubukcuğu mevcut; çoğunda t(8;21) ile ilişkilidir.
- M3 Akut promyelositik lösemi: %5-10: Çoğu hipergranüler promyelosit, çoğunda Auer cisimciği, hastalar daha genç (ortalama yaş 35-40); DIk insidansı yüksek; çoğunda t(15;17)
- M4 Akut myelomonositik lösemi: %15-20.Myelositik ve monositik diferansiasyon; myeloid elemanlarda değişken matürasyon; monoblastlar nonspesifik esteraz içerir. İnv(16) ile ilşkilidir.
- M5 Akut monositik lösemi: %10: M5a: kemik iliği ve kanda monoblast (peroksidaz -; nonspesifik esteraz+)ve promonositler M5b: periferi kanda matür monositler; her ikiside yaşlılarda sık, organomegali, lenfadenopati ve doku infiltrasyonu
- M6 Akut eritrolösemi: %5. Displastik eritrioid prekürsörleri ve non-eirtroid hücreler, <%30 myeloblastlar, ileri yaş, primer olguların %1’i; tedaviye sekonder gelişenlerşn %20’ si.
- M7 Akut megakaryositik lösemi: %1. megakaryosit seri balstaları baskın; trombosit spesifik antikorlar ile pozitif (GPIIb/IIIa yada vWF antikorları); myelofibrozis yada retikülin artışı
Morfoloji: Kemik iliğinde myeloid blastların %20 yada daha fazla oranda olması ile tanı konur. Birden fazla tipte blast görülebilir. Myeloblastlar 2-4 nükleollü, lenfoblastlara göre daha geniş sitoplazmalı hücrelerdir. Sitoplazmada azurofilik, peroksidaz pozitif granüller vardır. Auer cisimcikleri kırmızı boyanan, peroksidaz pozitif yapılar olup anormal azurofilik granüllerdir. Pek çok olguda bulunsa da, en sık t(15;17) ile (akut promyelositik lösemi) ilişkilidir. Myeloid diferansiasyonu gösterir. Monoblastlar kıvrıntılı yada multilobe nüveler içeir. Auer cisimciği içermezler. Peroksidaz negatif; nonspesifik esteraz pozitiftirler. Megakaryositik diferansiasyonda, fibrojenik sitokinlere bağlı kemik iliğinde fibrozisi gelişir. Eritroid diferansiasyon nadirdir. Nadiren periferik kanda blast görülmez (alösemik lösemi). Bu nedenle pansitopenik hastalarda, lösemiyi ekarte etmek için kemik iliği incelemesi yapılmalıdır.Klinik: Klinik bulgular ALL’ ye benzer. Haftalar yada birkaç ay içerisinde anemi, nötropeni ve trombositopeniye bağlı halsizlik, ateş, spontan mukozal yada kutanöz kanamalar görülür. En çarpıcı klinik bulgu, trombositopeniye bağlı oluşan kanamalardır. Kutanöz peteşi, ekimoz, serozal kanamalar, gingive ve üriner sistemde mukozal kanamalar sıktır. Akut promyelositik lösemide (M3), lösemik hücreler prokoagulan ve fibrinolitik faktörler üreterek kanama diatezini arttırırlar. Oral kavite, cilt, akciğer, böbrek, mesane ve kolonda mantar, pseudumonas ve diğer fırsatçılara bağlı enfeksiyonlar sıktır. Dokularda infiltrasyon daha nadirdir. Hafif lenfadenopati ve organomegali görülebilir. Monositik diferansiasyonda (M4, M5)cilt (lösemi kutis) ve gingiva infiltrasyonu görülebilir. ALL’ ye kıyasla SSS infiltrasyonu nadirdir. Çok nadiren kemik iliği yada periferik kan tutulumu olmadan kitle şeklinde bulgu verir (granülositik sarkom, meloblastoma, kloroma) ve birkaç yıl içinde sistemik AML’ ye dönüşür.
Prognoz: Tedavisi zordur. Kemoterapi ile %60 remisyon olsada, 5 yıllık hastalıksız dönem %15-30 olguda sağlanabilir. t(8;21) yada inv(16) le ilşkili olan olgularda prognoz nispeten iyidir. Önceden myelodisplastik sendromu olanlarda yada genotoksik tedavi alanlarda prognoz kötüdür. Yüksek riskli gruplar ve tekrarlama gösterenlere kemik iliği transplantasyonu uygulanır. T(15;17) ile ilişkili olanlarda vitamin A türevi retinoik asid, neoplastik promyelositlerin nötrofillere diferansiye olmasına yol açar. Nötrofillerin ömrü kısa olduğundan, kemik iliği temizlenir ve normal hematopoez için yer açılır. Tek başına uygulanırsa, tekrarlamalar sıktır.
Myelodisplastik Sendromlar
-
Myelodisplastik Sendromlar
İnefektif eritropoez ile ilişkili matürasyon defekti ve AML riski olan, klonal kök hücre hastalığıdır. Kemik iliği kısmen yada tamamen mutant multipotent kök hücreler ile doludur. Bu hücrelerin eritrosit, granülosit yada trombositlere diferansiasyon kapasitesi korunmuş olmakla birlikte inefektif yada düzensiz matürasyon mevcuttur. Periferik kanda sitopeni gelişir.- İdiopatik yada primer olan 50 yaş üzerinde ve yavaş gelişir.
- Tedaviye bağlı gelişen (t-MDS) genotoksik ilaç yada radyoterapiye sekonder olarak, tedaviden 2-8 yıl sonra gelişir. Tedaviye bağlı olgularda AML riski daha fazla olup daha çabuk gelişir.
Patogenez: Patogenezi bilinmez. Tanı anında kemik iliği genellikle hiperselüler olmakla birlikte, normo-hiposelüler olabilir. myelodisplastik progenitörlerde apoptozun artmış olması, inefektif eritropoez bulgusudur. Primer ve t-mds olgularında benzer kromozom anomalileri vardır: monozomi5, monozomi7, 5q ve 7q delesyonları, trizomi 8 ve 20q delesyonu.
Morfoloji: Lenfoid seri dışındaki tüm serilerde displastik diferansiasyon görülür. Halkalı sideroblastlar, Prusya mavisi ile boyanırsa eritrositlerde demir içeren perinükleer granüller (mitokondriler), B12 yada folat eksikliğine benzeyen megaloblastoid matürasyon, nükleer tomurcuklanma anomalileri bulunabilir. Nötrofillerde sekonder granüllerde azalma, toksik granülasyon ve/veya Döhle cisimcikleri görülebilir. Yalnız iki nükleer lob içeren nötrofiller (Paeudo-Pelger-Hüet hücreleri) sık olup nükleer segmentasyon içermeyen nötrofiller bulunabilir. Tek nükleer lob yada çok sayıda ayrı nüve içeren megakaryositler karakteristiktir. Myeloblastlar artmış olsa da, hücrelerin %20’ sinden azını oluşturur. Periferik kanda pseudo-Pelger-Hüet hücreleri, dev trombositler, makrositler, poikilositler, nispi yada tam monositoz belirlenir. Periferik lökositlerin %10’undan azı myeloblastlardan oluşur.
Klinik: Primer MDS 60 yaş üzerinde, güçsüzlük, enfeksiyonlar, kanamalar ile bulgu verir. Yaklaşık yarısında, rutin incelemeler sonucunda tanı konur. Kemik iliği yada kanda çok sayıda blast varlığı, ağır sitopeni, yüksek AML riski ve kötü prognoz ile ilişkilidir. Kromozomal anomali ve sitopeninin derecesi bağımsız risk faktörleridir. Ortalama sağkalım 9-29 ay olup iyi prognozlu olgular 5 yıl yada daha fazla yaşar. %10-40 olguda AML gelişir. T-MDS daha kötü prognozlu olup ortalama sağkalım 4-8 aydır. Genç hastalarda kemik iliği transplantasyonu yaralı olabilir. Yaşlılarda antibiotik ve transfüzyonlar ile destek tedavi uygulanır.
Kronik Myeloproliferatif Hastalıklar
Neoplastik hücre multipotent progenitör hücre olup matür eritrosit, trombosit, granülosit, monosit ve bazı olgularda lenfositleri oluşturabilir. Kronik myeoid lösemide ise, pluripotent kök hücre lenfoid ve myeloid seriyi oluşturur. En sık görülen 4 hastalık: 1) Kronik myeloid lösemi, 2) Polisitemia vera, 3) Esansiyel trombositoz, 4) Primer myelofibroz.. Neoplastik kök hücreler dolaşımda gezebilir; dalak gibi organlara yerleşip, ekstramedüller hematopoez odakları oluşturabilir. Hemen tümünde splenomegali mevcuttur. Kemik iliğinde fibrozis ve sitopeni görülebilir. Tümü zamanla akut lösemiye dönüşebilir.patolojik bulgular spesifik olmayıp klinik ve laboratuar bulguları ile birlikte değerlendirilir. KML olgularında belirlenen Philadelphia kromozomu (Ph), diğer kronik myeloproliferatif hastalıklarda görülmez.Kronik Myeloid Lösemi
Daha çok 25-60 yaş arası erişkinlerde görülüp 4. ve 5. on yılda pik yapar. Diğer kronik myeloproliferatif hastalıklardan belirli bir moleküler anomali ile ayrılır: 9. kromozomda yerleşen ABL geni ve 22. kromozomda yerleşen BCR genini çeren translokasyon (Ph). Sonuç olarak oluşan BCR-ABL füzyon geninin ürettiği füzyon proteini, tirozin kinaz aktivitesine yol açar. Hücre çoğalması artar, apoptoz azalır. Düzensiz myeloproliferasyon oluşur. %90 olguda Ph kromozomu (t(9;22) belirlenir.Morfoloji: Normal kemik iliği %50 hücre, %50 yağdan oluşur. KML’ de %100 hücreseldir. Hücrelerin çoğu olgunlaşmakta olan granülosit prekürsörleridir. Megakaryositler artmış olup bir kısmı küçük displastik yapıdadır. Eritrosit progenitörleri normal yada azalmıştır. Karakteristik bir bulgu buruşuk , yeşil-mavi sitoplazmalı histiositlerdir (deniz mavisi histiositler). Retikülin lifleri artmıştır. Periferik kanda lökositoz genellikle 100,000/mm3 ün üzerindedir. Dolaşan hücrelerin çoğu nötrofil, metamyelosit, ve myelositler olup myeloblastlar %10’ un altındadır. Eozinofili ve bazofili sıktır. Erken dönemde %50 trombositoz mevcuttur. Splenik kırmızı pulpada neoplastik ekstramedüller hematopoez nedeni ile belirgin splenomegali ve fokal enfarktüs sıktır. Hepatomegali ve hafif lenfadenopati belirlenebilir.
Klinik: Sinsi başlar. Hafif-orta derecede anemi, hipermetabolizm, çabuk yorulma, kilo kaybı, halsizlik ve iştahsızlık vardır. Bazen ilk bulgu splenomegaliye bağlı karında gerginlik yada splenik enfarktüse bağlı sol üst kadran ağrısı olabilir. Tedavi edilmese bile, 3 yıllık sağkalım mümkündür. 3 yıl kadar sonra %50 olgu hızlanma fazına geçer. Anemi ve trombositopeni artar; bazen belirgin bazofili oluşur. 6-12 ay içerisinde bu faz yerini akut lösemi benzeri bir tabloya bırakır (blast krizi). Geri kalan %50 olguda, hızlanma fazı olmadan blastik kriz gerçekleşir. Blastik krizde, %70 myeloblastlar görülür. Geri kalanın çoğunda Tdt, CD10 ve CD19 içeren B hücre belirleyicileri mevcuttur. Nadiren T blastik hücreler görülür. BRC-ABL kinaz inhibitörü olan ilaçlar %90 remisyon sağlar ama blastik krizler engellenemez. Blastik krizde, erken dönemde tedaviye yanıt versede; rekürrens çabuk olup tedaviye dirençli hale dönüşebilir. Bu nedenle, gençlerde, stabil dönemde uygulanan allojenik kemik iliği transplantasyonu en etkili tedavi yöntemidir. Uygun donör bulunduğunda, iyileşme oranı %75 civarındadır.
Polisitemia Vera
Kemik iliğinde eritrod, granülositik ve megakaryositik elemanların artışına yol açan multipotent myeloid kök hücre neoplazisidir. Eritrositoz (polisitemi), granülositoz ve trombositoza yol açar. Serum eritropoetin düzeyleri çok düşüktür. Kemik iliği hiperselüler olsada, bir miktar yağ dokusu mevcuttur. %10 olguda retikülin lifleri artmıştır. Erken dönemde konjesyona bağlı hafif organomegali belirlenir. Periferik kanda bazofiller artmış olup anormal büyük trombositler görülür. Geç dönemde kemik iliğinde yaygın fibrozis, dalak ve karaciğerde ekstramedüller hematopoeze bağlı belirgin organomegali gelişir. AML’ ye dönüşüm nadirdir.Klinik: 60 yaş civarında sıktır. Eritrosit ve hematokrit artışına bağlı bulgular oluşur. Hematokrit artışı genellikle kan volümünde artış ile birliktedir. Venöz dolaşım genişler. Hastalar pletorik ve siyanotiktir. Baş ağrısı, hipertansiyon, gastrointestinal semptomlar sıktır. Bazofillerden açığa çıkan histamine bağlı kaşıntı ve peptik ülser gelişebilir. Anormal kan akımı ve trombosit fonksiyonuna bağlı kanama ve trombüsler gelişir. %25 olguda ilk bulgu derin venöz trombüsler, myokard enfarktüsü ve inmedir. Ayrıca Buu-Chiari sendromu, portal ve mezenterik trombüslere bağlı barsak enfarktüsü, beyin venöz sinüslerinde trombüse bağlı hemorajik inmeler görülebilir. Burun ve diş eti kanamaları sık olup %5-10 olguda hayatı tehdit eden kanamalar oluşur.
LANGERHANS HÜCRELİ HİSTİOSİTOZ
-
LANGERHANS HÜCRELİ HİSTİOSİTOZ
Dendritik yapıdaki Langerhans hücreleri HLA-DR, S-100 ve CD1a eksprese ederler. Belirgin, vakuollü sitoplazma ve yarık yada katlantı içeren veziküler nüveleri vardır. Sitoplazmada Birbeck granülleri karakteristiktir.
Multifokal multisistem Langerhans hücreli histiositoz (Letterer-Siwe hastalığı) en sık 2 yaş altında görülür. sebooreik döküntülere benzeyen cilt lezyonları, hepatosplenomegali, lenfadenopati, pulmoner lezyonlar ve sonunda destrüktif kemik lezyonları gelişir. Kemik iliği infiltrasyonu pansitopeni, tekrarlayan otitis media ve mastoidit gibi enfeksiyonlar görülebilir. Tedavi edilmez ise öldürücüdür. Yoğun kemoterapi ile 5 yıllık sağkalım %50’ dir.
Unifokal unisistem Langerhans hücreli histiositoz (eozinofilik granülom) en sık kemik iliğini tutan, genişleyen eroziv lezyonlar olarak görülür. Histiositler eozinofil, nötrofil, lenfosit ve plazma hücreleri ile karışıktır. Eozinofiller belirgindir. Her kemik tutulabilirse de, en sık kafa kemikleri, kotsalar ve femurda yerleşir. Nadiren cilt, akciğer yada mide de yerleşir.Unifokal lezyonlar büyük çocuk yada erişkinlerde, iskelet sistemine yerleşir. Asemptomatik olabilir yada ağrı, hassasiyet ve patolojik kırığa yol açar. Spontan olarak gerileyebileceği gibi, lokal eksizyon yada radyoterapi ile iyileşir.
Multifokal unisistem Langerhans hücreli histiositoz genellikle küçük çocuklarda, multifokal, yumuşak dokuya uzanan, eroziv kemik defekteleri yapar. Hastaların yaklaşık %50’sinde arka hipofiz sapı tutulumu sonucunda diabetes insipitus gelişir. Kalvarial kemik defektleri, diabetes insipitus ve ekzoftalmus kombinasyonuna Hand-Schüller-Christian triadı denir. Çoğu hastada spontan gerileme olur. Diğerleri kemoterapi ile başarılı bir şekilde tedavi edilir.
TİMUS
Histoloji : Timus mediastende yerleşen lenfoepitelyal bir organdır. Lenfositleri mezodermden, epitel hücreleri endodermden (3-4. Faringeal keseler) gelişir. Parankimin içine girip onu lobullere ayıran bir kapsül ile çevrilidir. Her lobulün çevresinde koyu boyanan korteks ve merkezde açık boyanan medulla bulunur. Korteks T lenfositleri, epitelyal retiküler hücreler ve az sayıda makrofajlardan oluşur. Embrionik dönemde ve prepubertede lenfositler çok artar. Korteksde lenfositlerin çoğu ölür; kalanlar medullaya geçip dolaşıma karışır ve lenfoid dokulara ulaşılır. Medullada epitelyal retiküler hücreler, lenfositler ve yanı sıra keratohyalin granülleri içeren konsantrik dizilimli epitel hücrelerinin oluşturduğu Hassall korpüskülleri vardır. Timus T lenfositlerinin olgunlaşma yeridir.
Hiperplazi : Normal timusta lenfoid foliküller yoktur. Hiperplazide medullada lenfoid foliküller oluşur. Myastenya gravis, SLE ve romatoid artrit gibi otoimmun hastalıklarda çoğunlukla timusda hiperplazi oluşur. Myastenya graviste timusun myoid hücrelerine karşı duyarlılık kazanan T hücreleri lenfoid foliküllerde B hücrelerini uyararak antikor oluşumuna yol açar. Bunun sonucunda nöromuskuler bileşkedeki asetil kolin reseptörlerine karşı otoimmun reaksiyon gelişir.
Timoma
Timustaki epitelyal hücrelerin tümörleridir. Tümörde bulunan lenfositler normal yapıda olup neoplazik değildir.Beniğn Timoma : Sitolojik ve biolojik olarak beniğndir. Medulladaki epitel hücrelerine benzer hücreler oluşturur. Timomaların %60-70’ini oluşturur.
Malign Timoma : Tip I : Sitolojik olarak beniğn görünsede lokal invazyon ve nadiren uzak metastaz yapabilir. Timomaların %20-25’inin oluşturur.
Tip II (Timik Karsinom) :Hem sitolojik olarak hemde biolojik olarak maligndir. Timomaların %5’ini oluşturur. Çoğu iyi veya az diferansiye skuamöz karsinomdur. Daha az olarak görülen lenfoepitelyoma da olgun görünümlü lenfositlerde tümöre eşlik eder. Bir kısmında EBV varlığı belirlenmiştir. Bu açıdan nazofaringeal karsinoma benzer.
Timomalar orta yaşta sıktır. Myastenya gravisli olguların %15-20’sinde timoma bulunur. Tümör çıkarıldığında nöromuskuler bozukluk geriler. Hipogamaglobulinemi, SLE, saf eritrosit aplazisi ve nontimik kanserlerde timoma ile ilişkili olabilir. DALAK
-
DALAK
Histoloji
Dalak bağ dokusundan oluşan bir kapsülle çevrilidir. Kapsülden parankim içine uzanan trabekuller dalağı bölümlere ayırır. Dalakda lenfoid nodüllerin yer aldığı beyaz pulpa ve kandan zengin kırmızı pulpa bulunur. Kırmızı pulpa sinüzoidlerin arasında kordonlar (Billroth kordonları) şeklinde yerleşmiştir. Parankim içinde beyaz pulpada santral arterlerin çevresinde çoğu lenfositlerden oluşmuş bir kılıf vardır. Lenfoid nodüller B lenfositlerden oluşur. Kapillerden çıkan kan kırmızı pulpa hücrelerinin arasından geçerek sinüzoidlere gireraçık dolaşım. Kırmızı pulpada retiküler hücreler, makrofajlar, lenfositler, plazma hücreleri ve kan elemanları (eritrosit, trombosit, granulositler) bulunur. Primer tümörleri fibrom, osteom, kondrom ve en sık görülen lenfanjiom ile hemanjiomdur. Hematopoetik tümörler dalağı sık olarak tutar.Splenomegali :
Çoğu olguda splenomegali başka bir bölgedeki primer hastalığa sekonder olarak gelişir. Eritrosit, trombosit ve lökositler dalakta yıkılabilir.
İleri Derecede Splenomegali (1000 gm )- Kronik Myeloproliferatif hastalık (KML, Myeloid metaplazi)
- KLL (daha az oranda)
- Hairy cell lösemi
- Sıtma
- Gaucher hastalığı
- Lenfomalar
- Primer dalak tümörleri (nadir)
Orta Derecede Splenomegali (500-1000 gm)
- Kronik konjestif splenomegali (portal hipertansiyon veya splenik ven obstrüksiyonu)
- Akut lösemi
- Herediter Sferositoz
- Talasemi mayor
- Otoimmun hemolitik anemi
- Amiloidoz
- Niemann-Pick hastalığı
- Langerhans hücreli histiositoz
- Kronik splenit (özellikle enfektif endokardit ile birlikte)
- Tüberküloz, sarkoidoz, tifo
- Metastatik karsinom veya sarkom
Hafif derecede Splenomegali ( 500 gr. altında)
- Akut splenit
- Akut splenik konjesyon
- Enfeksiyöz mononükleoz
- Septisemi, SLE, intra-abdominal enfeksiyonlar gibi akut febril hastalıklar.
Dalak büyümelerinde hipersplenizm oluşabilir> anemi, lökopeni, trombositopeni